Reviews:
Cell Stress, Vol. 2, No. 8, pp. 184 - 199; doi: 10.15698/cst2018.07.147
Mitochondrial dysfunction and its role in tissue-specific cellular stress

1 Department of Cellular Biochemistry, University Medical Center Göttingen, Germany.
Keywords: mitochondrial dysfunction, cellular stress, mitochondrial pathology, therapy.
Received originally: 26/04/2018 Received in revised form: 13/06/2018
Accepted: 14/06/2018
Published: 13/07/2018
Correspondence:
David Pacheu-Grau, University Medical Center Göttingen, Department of Cellular Biochemistry, Humboldtallee 23, 37073 Göttingen, Germany. Phone: +49-(0)551-394571 David.Pacheu-Grau@med.uni-goettingen.de
Markus Deckers, University Medical Center Göttingen, Department of Cellular Biochemistry, Humboldtallee 23, 37073 Göttingen, Germany. Phone: +49-(0)551-395983 Markus.Deckers@medizin.uni-goettingen.de
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: David Pacheu-Grau, Robert Rucktäschel and Markus Deckers (2018). Mitochondrial dysfunction and its role in tissue-specific cellular stress. Cell Stress 2(8): 184-199. doi: 10.15698/cst2018.07.147
Abstract
Mitochondrial bioenergetics require the coordination of two different and independent genomes. Mutations in either genome will affect mitochondrial functionality and produce different sources of cellular stress. Depending on the kind of defect and stress, different tissues and organs will be affected, leading to diverse pathological conditions. There is no curative therapy for mitochondrial diseases, nevertheless, there are strategies described that fight the various stress forms caused by the malfunctioning organelles. Here, we will revise the main kinds of stress generated by mutations in mitochondrial genes and outline several ways of fighting this stress.
MITOCHONDRIA AND CELL METABOLISM
Mitochondria play a pivotal role in eukaryotic metabolism. They catabolise redox equivalents, derived from nutrient uptake, and use them to provide the bulk of cellular energy in the form of ATP. The oxidative phosphorylation system (OXPHOS) is responsible for this energy production and it is composed of five multi-oligomeric complexes present in the inner mitochondrial membrane. Transfer of electrons through complexes I to IV reduce molecular oxygen to water. This process is coupled to proton pumping from the matrix to the intermembrane space (IMS), while the return of protons to the matrix through the F1Fo ATPase generates ATP [1]. However, an inefficient flow of electrons through the respiratory chain complexes would partially reduce oxygen and produce reactive oxygen species (ROS) like superoxide and hydrogen peroxide. At low concentrations, these molecules act as second messengers and can activate gene transcription and trigger cellular responses, like cellular growth, production of cellular antioxidants or stimulation of mitochondrial biogenesis [2][3]. However, once a certain threshold is exceeded, these molecules may incite oxidative damage in the form of mitochondrial DNA (mtDNA) alterations or lipid peroxidation, generating cellular stress that leads to aging or cell death.
–
In addition, mitochondria are involved in many other key cellular functions. Dissipation of the proton gradient by uncoupling proteins (UCPs) generates heat instead of energy and this plays an important role in exposure to cold or hibernation [4]. Calcium (Ca2+) uptake inside mitochondria is mediated by the mitochondrial calcium uniporter (MCU). Although the complex has a low affinity for Ca2+, the transport takes place due to the high concentration of Ca2+ (>10 µM) present in micro domains located in the contact sites between endoplasmic reticulum (ER) and mitochondria [5]. The mitochondrial Ca2+ uptake not only shapes the cytosolic Ca2+ dynamics, which is crucial for muscle contraction, exocytosis and gene transcription, but also modulates at least three dehydrogenases of the Krebs cycle, thus regulating energy metabolism. Finally, Ca2+ overload in mitochondria regulates apoptosis due to formation of the permeable transition pore (PTP) and release of cytochrome c from the IMS [6]. Mitochondria are involved in the biogenesis and maturation of different cofactors, like heme, biotin or iron-sulfur (Fe/S) clusters. Despite the chemical simplicity of Fe/s clusters, their biosynthesis requires more than two dozen proteins in eukaryotes and takes place both in mitochondria and the cytosol [7]. Alterations in these mechanisms are linked to severe neurodegenerative, metabolic or haematological diseases [8].
–
Since mitochondria take part in many different metabolic processes, mitochondrial malfunction can affect numerous aspects of the cell. As a consequence, various forms of cellular stress are generated, leading to a large variety of pathological conditions. Here, we review different forms of cellular stress caused by mitochondrial malfunction and the strategies used to fight this stress.
MITOCHONDRIAL DEFECTS
Mitochondria have retained their own genome, the mtDNA. This small, circular, double-stranded DNA is located in the mitochondrial matrix in all cell types, and can be found with copy numbers that range from several to thousands of copies. In human, the mtDNA encodes 13 polypeptides of the respiratory chain, as well as for part of the translation machinery, required for the synthesis of these polypeptides within mitochondria: two ribosomal RNAs (mt-rRNAs) and 22 transfer RNAs (mt-tRNAs) [9]. The remaining mitochondrial proteins (approx. 99%) are encoded in the nucleus, synthesized on the cytosolic ribosomes and imported into mitochondria. Therefore, we will distinguish between mitochondrial malfunction caused by mutations in the mtDNA and those caused by mutations of nuclear genes encoding mitochondrial proteins.
–
Alterations in the mtDNA
Some features of mtDNA make it especially sensitive to oxidative damage and mutation. Firstly, mtDNA has no introns, so every single nucleotide carries information essential for protein coding; mtDNA is naked, there are no histone proteins protecting it from damage; and although DNA repair systems do exist in mitochondria, their mechanisms and extent are poorly understood [10], therefore mutations usually remain and are transmitted to the next generation until they are removed by selection [11]. Moreover, the proximity to the respiratory chain, a ROS producing source, increases the risk of potential damage. For all these reasons, the mutational rate of the mitochondrial genome is much higher than that of the nuclear genome[12].
–
 | FIGURE 1: Mitochondrial dysfunctions are related to mutations in mtDNA and defects in nuclear encoded mitochondrial proteins. (I) Overview of mutations within the mtDNA. (II) The majority of mitochondrial defects based on a malfuntion of OXPHOS complexes and mitochondrial translation. (III) Defects in other processes, like mitochondrial fusion and fission or lipid homeostasis, leads to different mitochondrial diseaes. (IV) Different strategies to figth the diverse forms of mitochondrial stress. (a: Leigh Syndrome LS; b: Leber Hereditary Optic Neuropathy LHON; c: Neurogenic Muscle Weakness, Ataxia and Retinitis Pigmentosa NARP; d: Mitochondrial Encephalomyopathy, Lactid Acidosis and Stroke-like Episodes MELAS; e: Myoclonic Epilepsy and Ragged Red Fiber Disease MERRF; f: Sensorineural Hearing Loss SNHL; g: mitochondrial non-syndromic Hearing Loss; D: Kearns Sayre Syndrome KSS). |
–
Pathological changes in the mtDNA can appear as point mutations in protein coding sequences, mt-tRNAs or even mt-rRNAs. In addition, major rearrangements of mtDNA, like deletions or insertions/duplications, are a cause of disease. Due to the fact that every cell contains a variable number of mtDNA molecules, mutations can be present in homoplasmy (all copies share the same mtDNA genotype) or heteroplasmy (only a population of DNA is mutated). The level of heteroplasmy of a mutation is a critical determinant of the cellular stress of a certain tissue or organ and has a major role in the disease phenotype. Finally, a reduction of mtDNA copies (depletion syndrome) can also hamper energy production and generate cellular stress (see Table 1, Figure 1) [12].
–
TABLE 1. Types of mitochondrial disease caused by mitochondrial encoded genes. |
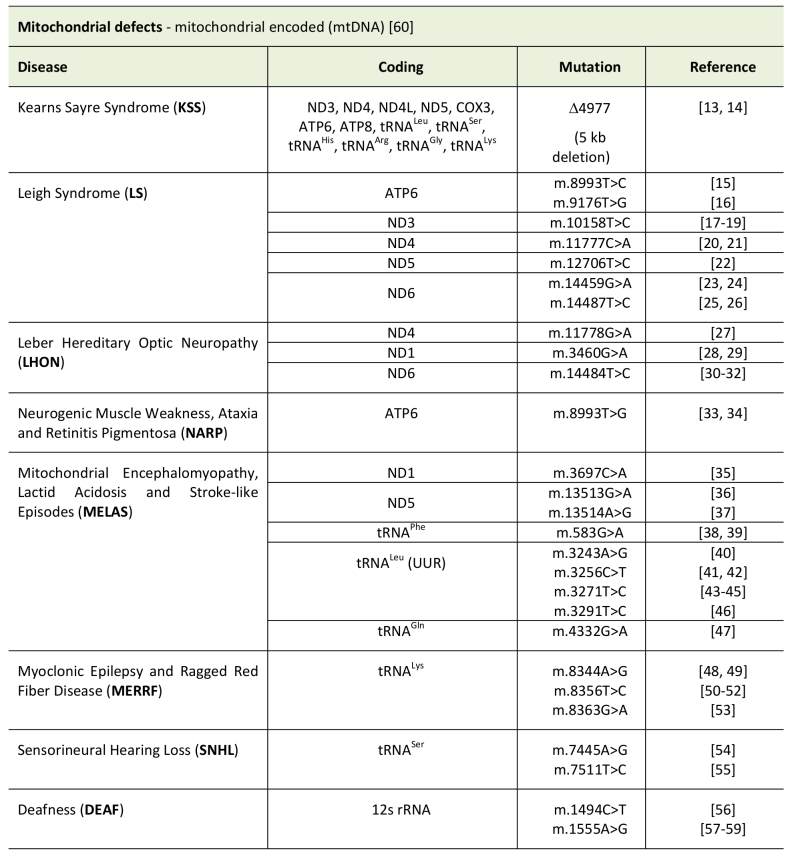 |
| [13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35] [36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60] |
–
Alterations in the mitochondrial proteins encoded in the nucleus
Due to the diverse cellular roles that mitochondria fulfill, there are many mitochondrial processes that cause a pathology when disturbed. In the last years, massive sequencing approaches have significantly increased the number of known mutations implicated in mitochondrial diseases. Examples of this are defects in: factors involved in the biogenesis or integrity of respiratory chain complexes, those that regulate mtDNA maintenance, proteins required for transcription of mt-mRNA elements involved in translation of mtDNA encoded proteins, regulators of lipid metabolism, factors involved in cellular signalling and even enzymes of the Krebs cycle (see Table 2).
–
TABLE 2. Mitochondrial defects caused by nuclear encoded genes. |
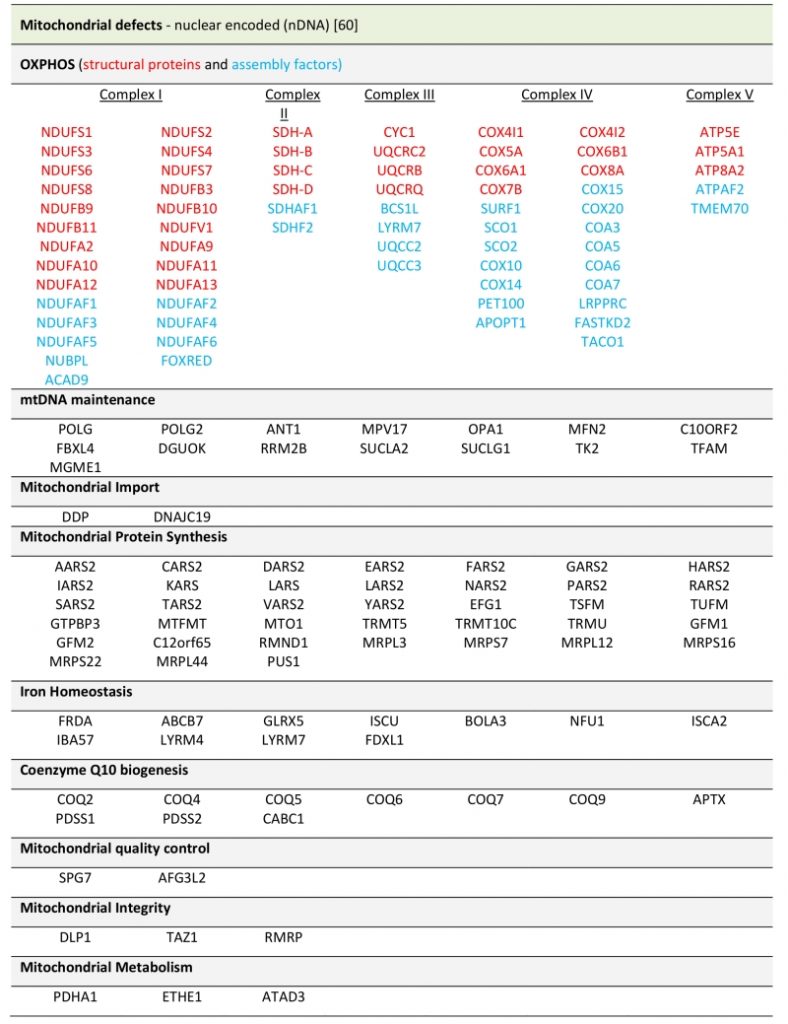 |
| [60] |
CELLULAR EFFECTS ON DIFFERENT TISSUES
Typically, mitochondrial disorders have been divided between those presenting with multiple symptoms, usually known as syndromes, and those characterized by tissue specific phenotypes. It remains to be addressed, which factors determine the tissue-specificity of mitochondrial diseases. However, to better address the different kinds of stress caused by mitochondrial distress, we will describe them classified by tissues/organs and give some examples of alterations that cause these problems.
–
Sensory organs
Hearing loss is one of the most prevalent sensory disorders [13]. Genetic factors are thought to account for more than half of congenital and childhood-onset hearing loss, including mutations in mtDNA [14] and mitochondrial nuclear genes like the heme A biogenesis factor COX10 [15][16] or the AAA protease responsible for complex III assembly BSC1L [17][18].
–
Mutations in the 12S mt-rRNA (m.1555A>G and m.1494 C>T) have been associated with aminoglycoside-induced ototoxicity and mitochondrial non-syndromic hearing loss. Studies using mitochondrial cybrids derived from Hela cells and lymphoblasts have shown that these mutations affect the integrity and fidelity of the mitochondrial ribosome, therefore causing decreased mitochondrial translation, either in the presence or the absence of aminoglycosides, and resulting in a cell growth defect [19][20]. However, a study using osteosarcoma 143B derived cybrids showed no effect on mitochondrial translation after aminoglycoside treatment [21]. This discrepancy, the phenotypic differences between asymptomatic relatives and patients all harbouring the same mutational load and the fact that only in some cases the defect arose upon antibiotic treatment, raised the search for modifying factors of aminoglycoside induced ototoxicity within the nuclear genetic background [22]. Indeed, no negative effect was observed after aminoglycoside treatment in primate cells from the Cercophiteciade family where the m.1494 C>T was fixed as the wild-type allele and cells carried a compensating mutation in mitochondrial ribosomal protein S12 (MRPS12) [23], whereas primate cells from orangutan carrying the m.1555A>G mutation and no MRP mutation showed a drastic effect after antibiotic treatment [24]. In addition, this biochemical effect has been linked to stress signalling. Cybrids carrying the m.1555A>G mutation showed hypermethylation of the mitochondrial ribosome, disturbed mitochondrial translation and assembly of the respiratory chain, resulting in increased production of ROS. Enhanced superoxide levels are sensed by AMPK, which signals further to E2F1, activating pro-apoptotic signalling in the cell. This induction seems to be tissue-specific, happens mainly in the inner ear and may explain the specific hearing defect observed in the presence of this particular mutation [25] (see Table 1).
–
Eye complications are also frequently found to be associated with mitochondrial dysfunction [26] and can be divided into primary and secondary. Primary afflictions are caused by genetic defects, whereas secondary afflictions are produce by hypertensive angiopathy of the retinal arteries, or diabetic retinopathy in mitochondrial diseases with diabetes [27]. Mitochondrial optic neuropathies have been associated with mutations in mtDNA and in nuclear genes. The most frequent eye disorder due to mtDNA mutation is Leber´s hereditary optic neuropathy (LHON) [28]. LHON usually affects young male adults and is characterised by mostly bilateral subacute or acute, painless, loss of central vision, with decreased colour vision [29]. There are three main mtDNA mutations that underly the majority of LHON cases and all of them are found in complex I genes: m.11778G > A in the ND4 gene, m.3460G > A in the ND1 gene, and m.14484T > C in the ND6 gene (see Table 1). In addition, these mutations are usually present in homoplasmy, indicating that probably other factors are involved in the development of the disorder. The molecular mechanisms underlying LHON are not yet fully understood. There have been some risks factors proposed, like specific mitochondrial haplogroups, smoking, alcohol consumption, and the use of some antibiotics. Differences in mitochondrial mass have been also postulated to play a role in the incomplete manifestation of the disease. LHON mutation carriers with no pathological phenotype have significantly higher mtDNA copy number in leukocytes than affected carriers. By comparing fibroblasts from unaffected and affected mutation carriers, along with controls, it was shown that unaffected carriers have increased mitochondrial transcripts, respiratory chain proteins and enzyme activities compared to controls and affected carriers. Therefore, increased mitochondrial mass may play a protective role in LHON and compensate for complex I dysfunction [30]. In addition, males seem to be more affected because of the lack of protective effects from estrogen. Indeed, a study using cybrids carrying LHON mtDNA mutations showed that the addition of estradiol increased mitochondrial biogenesis and decreased ROS production by enhancing the activity of detoxifying enzymes like SOD2, leading to a decrease in apoptosis [31] (see Table 1).
–
The most common eye afflictions associated with nDNA mutations are autosomal dominant optic atrophy (ADOA), most frequently due to mutations in the Dynamin-like GTPase OPA1, and autosomal recessive optic atrophy (AROA), which has been mainly associated with mutations in the aconitate hydratase ACO2, or the uncharacterised transmembrane protein TMEM126A (OPA7) [28]. ADOA is clinically characterised by bilaterally symmetric progressive deterioration of the central visual acuity. Approximately 60-70% of ADOA cases are caused by genetic alterations in OPA1, other genes implicated in this pathology are OPA2 [32], OPA3 [33], OPA4 [34], OPA5 [35], OPA8 [36] and WFS1 [37] (see Table 1). OPA1 is a protein with eight different isoforms, processed by the mitochondrial metallochaperones YME1L and OMA1 [38][39][40]. The best-known function of OPA1 is for inner mitochondrial fusion during mitochondrial dynamics. In addition, OPA1 is involved in the remodelling of cristae by tethering inter-cristae membranes and proper function of the protein is required for maintaining cristae structure [41]. OPA1 mutations cause defective mitochondrial fusion and altered cristae structure, leading to direct effects on mitochondrial bioenergetics, including a decreased mitochondrial membrane potential and ATP synthesis and increased ROS production [42]. Interestingly, deletion of YME1L in murine heart, which alters OPA1 processing and function in a tissue-specific way, causes dilated cardiomyopathy (Figure 1) [43].
–
AROA presents with progressive impairment of visual capacity. The defect could either be spontaneously recovered or may lead to bilateral and progressive blindness [29]. Mutations in ACO2, affect the mitochondrial tricarboxylic acid cycle and therefore mitochondrial energy supply is depleted in patients [44]. Although there have been several AROA patients with mutations in TMEM126A, the exact function of the protein and therefore the molecular mechanism underlying optic atrophy has yet to be determined [45][46][47].
–
Heart
Cardiac muscle has a high energetic demand, therefore cardiac complications are frequent among mitochondrial diseases. One of the most common cardiac afflictions present in these pathologies is cardiomyopathy, which is estimated to occur in 20-40% of children with mitochondrial disease [4][48][49]. However, other symptoms like arrhythmia, conduction defects, pulmonary hypertension, dilated aortic root, pericardial effusion or coronary heart disease can also be developed as consequence of mitochondrial malfunction [5][50].
–
Mitochondrial cardiomyopathies are characterised by abnormal myocardial structure or function that results from genetic defects that impair the mitochondrial respiratory chain [6][50]. Hypertrophic cardiomyopathy is the most common form, present in more than 50% of cases [7][48], but other forms, like dilated, restrictive, histiocytoid and left ventricular non-compactation cardiomyopathies can also be found among these patients [8][51].
–
As described before (see above), genetic defects affecting the integrity of respiratory chain complexes, mitochondrial translation, maintenance of mtDNA, lipid metabolism and other metabolic pathways inside mitochondria might lead to cardiac disease. Several important perturbations have been described in subunits or factors required for the proper assembly of respiratory chain complexes. In general, mutations in these proteins cause an impairment of respiration and ATP production, increased ROS production and finally, cellular stress derived from a bioenergetics impairment. To date, pathological mutations have been found in 26 structural subunits of complex I [9][52] , that together with mutations in assembly factors represent around 30% of childhood mitochondrial diseases [11][53]. Complex I defects can be present with isolated cardiomyopathy or together with multi-organic failure. Mutations in subunits of complex II or III have also been associated with different types of cardiomyopathy [12][54][55][56]. Of special interest are defects of the cytochrome c oxidase, caused by mutations in assembly factors and in nuclear-encoded structural subunits. Mutations in the complex IV assembly factors COX10 and COX15 have been associated with hypertrophic cardiomyopathy [16][57]. Both assembly factors are involved in the biosynthesis of heme A, the prosthetic group of the cytochrome c oxidase. Mutations in COX6B1, have been associated with cardiomyopathy and encephalopathy and showed decreased levels of the mature cytochrome c oxidase complex in patient-derived tissues and cells [12][58][59]. Although COX6B1 was thought to be a loosely interacting structural subunit of complex IV, studies have postulated Cox12 (yeast homolog of COX6B1) to be involved in the delivery of copper to Cox2, together with other metallochaperones like Sco1, Sco2 and Coa6 [13][60]. Indeed, mutations in human SCO2 have been mainly associated with cardioencephalopathy [14][61][62][63], whereas mutations in SCO1 have been associated with hepatic failure and encephalopathy [19][20][64][65], as well as cardiomyopathy [21][66]. Both proteins contain a CXXXC that is able to coordinate copper. They bind to apo-COX2 and deliver two copper atoms to the CuA center. Both enzymes have different but cooperative functions and disruptions in their function impair the maturation of cytochrome c oxidase[22][67]. In addition, the SCO1 and SCO2 proteins are involved in regulating cellular copper homeostasis [23][68]. Recently, it has been shown that SCO1 keeps the copper transporter CTR1 in the plasma membrane, this function being essential for the development of adult myocardium in mice [24][69]. Mutations in COA6 have been described in infants with hypertrophic cardiomyopathy and combined complex I and IV, or isolated complex IV deficiency in the heart [25][70][71]. COA6 is required for cytochrome c oxidase assembly [26][72][73]. It is involved in the insertion of copper into COX2 and it has been described to interact with SCO2 [27][74] and SCO1 [28][75] after the translocation of the COX2 C-terminal domain into the IMS by COX18 [76]. However, why disturbance of copper metabolism, or ultimately of the cytochrome c oxidase, specifically affects the heart remains unclear (see Table 2, Figure 1).
–
Mutations in the mitochondrial translation machinery have also been associated with cardiomyopathy. Primary defects produce a decreased synthesis of mitochondrial polypeptides, but ultimately also impair mitochondrial bioenergetics and cause cellular stress. Mutations in the 16S mt-rRNA, and the m.1555A>G mutation in the 12S mt-rRNA, have been associated with hypertrophic and restrictive cardiomyopathy [30][77][78]. Mutations in ribosomal proteins (MRPL3 and MRPL44) and the translation elongation factor (TSFM) can cause cardiomyopathy, together with multi-organic disease [31][79][80][81][82]. Finally, defects in mitochondrial tRNAs can be linked to isolated cardiomyopathy or multi-organic disfunction [83][84][85] (see Table 1 and Table 2).
–
Alterations of lipid metabolism inside mitochondria can also be a determinant for cardiac disease. Barth syndrome is an x-linked autosomal recessive disease, characterized by cardiomyopathy, skeletal myopathy, neutropenia, growth retardation, and 3-methylglutaconic acidurea [32][86][87][88]. This disorder is caused by mutations in the Tafazzin protein, TAZ1, a mitochondrial acyl-transferase involved in the biogenesis of cardiolipin (CL), a phospholipid almost exclusively found in the inner mitochondrial membrane [33][89]. The adequate presence of CL is required for structural stability of many critical protein complexes in the mitochondrial membrane and it is therefore essential for many mitochondrial processes ranging from protein import, cristae morphology, function of the respiratory chain or cell stress signaling [34][88]. Interestingly, oxidation of CL causes loss of interaction with cytochrome c, a pre-requisite for triggering apoptosis. Oxidized CL has been found to be involved in the opening of the mitochondrial permeability transition pore (MPTP). In addition, CL is exposed to the outer mitochondrial membrane during apoptosis, where it is used as a binding platform for pro-apoptotic factors. Therefore, CL homeostasis plays an important role in cardiomyocyte programmed death upon ischaemia or reperfusion (Figure 1) [35][88].
–
Mutations in another lipid related enzme, the acylglycerol kinase AGK, have been associated with hypertrophic cardiomyopathy, myopathy, cataracts, exercise intolerance and lactic acidosis (Sengers syndrome). AGK was recently described as a component of the carrier protein translocase of the inner membrane (TIM22) [36][90][91], meaning that a defective import of carrier proteins alters mitochondrial metabolism and may disturb the function of the heart.
–
Neurological disorders
Similar to previously described organs and tissues and due to the high energy demands, neurological complications are commonly linked to mitochondrial disfunction. Indeed, some of the most known mitochondrial syndromes caused by abnormalities in the mtDNA present with drastic neurological symptoms: Kearns–Sayre syndrome (KSS), a multisystem disorder with progressive external ophthalmoplegia, pigmentary retinopathy, heart block and frequently other signs like ataxia, dementia or endocrine problems is associated with single deletions of mtDNA [92]. MELAS (Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis and Stroke-like Episodes) is caused in 80% of the cases by the m.3243A>G mutation in the tRNALEU(UUR) gene, although there have been other mutations described in protein coding genes [93][94][95][32][88][96]. MERRF (Myoclonic Epilepsy and Ragged-Red Fiber), which usually also presents with cerebellar ataxia is mainly caused by mutations in the tRNAlys gene (m8344A>G, m8356T>C, m.8363G>A), being the m.8344A>G the most frequent of them [97][98][99][100]. The previously described defects affect the gene expression machinery of the mitochondrial genome and will generally affect mitochondrial protein synthesis, moreover an increased ROS production has been described in cybrids carrying the MELAS m.3243A>G mutation or the KSS associated common deletion Δ4977 [41][101]. In addition, there are mutations described in protein coding genes or in mitochondrial nuclear genes that would only affect individual complexes of the respiratory chain: NARP (Neuropathy, Ataxia and Retinitis Pigmentosa) has been mainly associated to the m.8933T>G/C mutation in the complex V subunit mt-ATP6 [102]. NARP patient derived cells were also found to have increased ROS production and decreased levels of ATP production [101]. Leigh syndrome is a progressive neurometabolic disorder that usually presents with seizures, hypotonia, fatigue, nystagmus, poor reflexes, eating and swallowing difficulties, breathing problems, poor motor function, and ataxia. This unique mitochondrial disorder is found to be caused by both mutations in the mtDNA and the nDNA. Mutations in many different genes have been identified to be the origin of Leigh syndrome, including mtDNA subunits of the complex I, IV and V, mt-tRNAs, nuclear encoded subunits of complex I, IV and II, the pyruvate dehydrogenase complex, or some assembly factors of the cytochrome c oxidase (SURF1, SCO1, SCO2, COX10, COX15) or complex III (BSC1L) [103]. Cells derived from patients with 3 different complex I mutations and Leigh syndrome exhibited increased ROS production [101][104] (see Table 1 and Table 2, Figure 1).
–
As already mentioned, many more genes are being identified as the reason behind mitochondrial dysfunction. Defects in mtDNA maintenance may result into defective mtDNA replication and lead to quantitative loss of mtDNA (mtDNA depletion) or qualitative one (mtDNA deletion). Downstream perturbation of mitochondrial protein synthesis will final lead to a bioenergetics defect. MPV17 is a mitochondrial inner membrane protein involved in maintenance of mtDNA. It is believed to be involved in the import of deoxynucleotides into mitochondria. Pathogenic variants in MPV17 have been reported to cause hepatocerebral mtDNA depletion syndrome with liver failure, de velopment delay and other neurological manisfestations [105][106]. In addition, infantile Navajo neuropathy (NNH), a neurohepatological disorder prevalently present among Navajo children in the southwestern of USA has been found to be caused by mutations in MPV17 [107]. A recent report analysing new pathological variants in MPV17 showed that most patients exhibited a single or combined respiratory chain complex activity decrease [108] (see Table 2).
–
Aminoacyl-tRNA synthetases (ARS), are a family of proteins encoded in the nucleus and present in either the cytosol or mitochondria that ensure the proper conjugation of an amino acid with its cognate tRNA molecules. All mt-ARS are synthesized in the cytosol, imported to mitochondria due to an N-terminal targeting sequence (presequence) which is cleaved upon translocation to the matrix. Pathogenic variants of mt-ARS will affect mitochondrial translation and have been implicated in human neurological disorders of the brain, spinal cord and motor neurons in addition to other symptoms. Some of the most typical presentations are leukoencephalopathy with involvement of the brainstem and spinal cord and high lactate due to mt-aspartyl-tRNA synthetase (DARS2) mutations [109], leukoencephalopathy with thalamus and brainstem involvement and high lactate, caused by mt-glutamyl- tRNA synthetase (EARS2 [110]). However, there are other mt-ARS mutations which may also produce white matter lesions. The similar symptoms shown by ARS mutations may imply a shared mechanism of disease, however such a mechanism has not been yet demonstrated. Among the possible molecular reasons are: a reduced aminoacylation activity, altered dimerization, mislocalization, gain of function though pathogenic interactions and loss of noncanonical function [111] (see Table 2).
NEW STRATEGIES TO FIGHT MITOCHONDRIAL DERIVED STRESS
Nowadays there is no actual treatment for mitochondrial diseases. Nevertheless, in the last years a number of therapeutic strategies have been proposed, mainly in animal models. They can be classified into those acting on common pathways, and therefore applicable to different diseases, and those which aim to ameliorate a particular disorder (Figure 1) [112].
–
Those tissues or organs affected by decreased ATP production, and therefore impaired bioenergetics, can benefit from increased mitochondrial mass and activity. The transcriptional co-activator peroxisome proliferator activated receptor-1alpha (PGC1alpha) is the master regulator of mitochondrial biogenesis. It increases the activity of several transcription factors, like the nuclear respiratory factors (NR1 and NR2), thereby controlling the expression of OXPHOS related genes. In addition, PGC1alpha interacts with the peroxisomal proliferator activator receptors (PPARs), which regulate the expression of fatty acid oxidation genes [113]. PGC1alpha is activated either by deacetylation by Sirt1, or phosphorylation by AMPK, both of which can be modulated pharmacologically [114].Under physiological conditions, PCG1alpha shows its highest expression levels in the heart, and mouse models lacking this protein have shown a normal cardiac function in unstimulated conditions. However, an impaired cardiac function was observed during certain stress conditions, like intense exercise or aortic constriction. Thus, the physiological role of PCG1alpha seems to be in fighting cellular stress [115].
–
Another possible strategy is to bypass the block in the respiratory chain from specific complex defects. In such a way, electrons would flow again and reduce ROS production. Concomitantly, unaffected complexes would pump protons across the inner membrane and increase ATP production. The yeast Saccharomyces cerevisiae NADH reductase (Ndi1), which transfers electrons from NADH to coenzyme Q (CoQ), has been used to bypass CI defects [116]. In a similar approach, the alternative oxidase (AOX), which transfers electrons from CoQ to molecular oxygen in different organisms, has been used to bypass CIII and IV defects in cell culture [117] and to ameliorate to different extent respiratory defects in fly models [118][119]. The enzyme has been successfully expressed in murine models [120], however correction of respiratory chain defects has not been shown yet in vivo in mammals.
–
As previously described (see above), the dynamin-like GTPAse OPA1 is required for proper mitochondrial shaping. Regulating fission and fusion helps fight mitochondrial malfunction. Increasing the expression of long isoforms of OPA1 improves respiration efficiency by enhancing supercomplex assembly and protects in vivo from many insults, such as ischemia/reperfusion, denervation/induced muscle atrophy, and OXPHOS deficiency [41][121][122].
–
In order to cope with increased oxidative damage generated in damaged mitochondria, different small molecules with antioxidant properties have been tested. Some examples, like Idebenone, lipoic acid, or Coenzyme Q10 , directly transfer electrons to the respiratory chain and bypass defective complexes. Others, like EPI-743 and RP103, enhance the biogenesis of glutathione, an important cellular antioxidant. KH176, can reduce altered cellular ROS levels and protect OXPHOS deficient cells against redox stress by targeting the Thioredoxin/Peroxiredoxin system [123]. MTP-131 is a member of the Szeto‐Schiller (SS) peptide family and binds to CL. It increases OXPHOS capacity and improves the way mitochondria respond to metabolic changes. L-Arginine, a donor of nitric oxide, which thus regulates vascular tone, was shown to induce an improvement in aerobic capacity and muscle metabolism in models for mitochondrial disease [124].
–
Finally, genetic approaches can be used to correct mutations at a genomic level. Mitochondrially targeted restriction endonucleases have been used to shift heteroplasmy levels in cell lines with mutations in mtDNA and in heteroplasmic mice. Introduction of TALE and zinc finger nucleases (TALEN and ZFN) enabled the addition of specificity to the nucleases so that mutant DNA molecules could be selected for by directing unspecific restriction enzyme (FokI) to appropriate specific sequence assembling ZFN or TALE modules [125][126]. However, this approach requires very large constructs that do not so easily fit into adeno-associated viruses (AVV) vectors. The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) system is a bacterial immune system that has been modified for genome engineering. Due to the simplicity and adaptability of this technique, CRISPR has quickly displaced the previously established TALENs or ZFNs for genome engineering. CRISPR consists of two elements: a guide RNA (gRNA) and a non-specific CRISPR-associated endonuclease (Cas9). The gRNA is a short synthetic RNA composed of a “scaffold”, necessary for Cas9 binding, and a 20 nt targeting sequence that is specific to the gene of interest [127]. CRISPR was originally employed to knock-out target genes, but it has also been used to chromosomally modify or tag proteins, and to activate or repress target genes. However, the viability of this approach to target mitochondrial genes, mainly because of the requirement of a reliable nucleotide import system into mitochondria is not yet clear [128].
CONCLUDING REMARKS
Mitochondrial diseases show very complex and various clinical presentations. Because of the dual genetic origin of mitochondrial proteins, the number of genes susceptible of causing a mitochondrial pathology is large. Thus, the diagnostic process of mitochondrial diseases is usually complicated and very long, and in many cases although there is a clear suspicion of a mitochondrial defect, the final defect behind the phenotype remains undercover [129].
–
Despite the heterogeneity of the diseases and the genetic defects the final kind of cellular stresses are similar. In the most cases, there is a bioenergetic defect or an increased production of ROS. It remains unclear why although many genetic defects are present in the whole body, only certain tissues are affected. There may be different mechanisms to cope with mitochondrial stress, which may be tissue specific [25]. Indeed, disease models of mtDNA replication machinery failure have been linked to imbalance of the cellular dNTPs pool and consequently to increased glutathione biogenesis through de novo serine biogenesis. This metabolic switch was proposed to be a specific and rapid response to cellular stress/mtDNA damage in skeletal muscle and heart [130]. Together with this pathway, transcriptional response and mitochondrial unfolded protein response constitute the integrated mitochondrial stress response (ISRmt), which is controlled by the metabolic signalling regulator mTORC1 in muscle. However, long-term activation of cellular stress responses may be detrimental since chronic upregulation of anabolism contributes to mitochondrial myopathy pathogenesis [131]. In addition, there is a certain multifactorial component in mitochondrial diseases. In some cases, ancient mt-rRNAs mutations rendered no adverse phenotype unless some environmental factors were used [24]. Therefore, the different incorporation and tolerance of tissues and organs for different xenobiotics may be different. Population variation plays also an important role in enhancing/ diminishing mitochondrial-related phenotypes, like population polymorphisms in mt-rRNAs and side effects derived from antibiotic treatment [132]. Nevertheless, a deeper investigation will be required to understand tissue specificity of mitochondrial diseases.
–
There have been several approaches proposed to treat mitochondrial diseases, however their application to the clinics is still a challenge [112]. Nevertheless, the discovery of new genome editing tools and the development of stem cell technologies will provide open new avenues of possibilities for the treatment of mitochondrial diseases.
References
- D. Mick, S. Dennerlein, H. Wiese, R. Reinhold, D. Pacheu-Grau, I. Lorenzi, F. Sasarman, W. Weraarpachai, E. Shoubridge, B. Warscheid, and P. Rehling, "MITRAC Links Mitochondrial Protein Translocation to Respiratory-Chain Assembly and Translational Regulation", Cell, vol. 151, pp. 1528-1541, 2012. http://dx.doi.org/10.1016/j.cell.2012.11.053
- E.D. Yoboue, and A. Devin, "Reactive Oxygen Species-Mediated Control of Mitochondrial Biogenesis", International Journal of Cell Biology, vol. 2012, pp. 1-8, 2012. http://dx.doi.org/10.1155/2012/403870
- S. Di Meo, T.T. Reed, P. Venditti, and V.M. Victor, "Role of ROS and RNS Sources in Physiological and Pathological Conditions", Oxidative Medicine and Cellular Longevity, vol. 2016, 2016. http://dx.doi.org/10.1155/2016/1245049
- R.A. Busiello, S. Savarese, and A. Lombardi, "Mitochondrial uncoupling proteins and energy metabolism", Frontiers in Physiology, vol. 6, 2015. http://dx.doi.org/10.3389/fphys.2015.00036
- E. Penna, J. Espino, D. De Stefani, and R. Rizzuto, "The MCU complex in cell death", Cell Calcium, vol. 69, pp. 73-80, 2018. http://dx.doi.org/10.1016/j.ceca.2017.08.008
- A. Raffaello, C. Mammucari, G. Gherardi, and R. Rizzuto, "Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes", Trends in Biochemical Sciences, vol. 41, pp. 1035-1049, 2016. http://dx.doi.org/10.1016/j.tibs.2016.09.001
- R. Lill, "Function and biogenesis of iron–sulphur proteins", Nature, vol. 460, pp. 831-838, 2009. http://dx.doi.org/10.1038/nature08301
- M. Cardenas-Rodriguez, A. Chatzi, and K. Tokatlidis, "Iron–sulfur clusters: from metals through mitochondria biogenesis to disease", JBIC Journal of Biological Inorganic Chemistry, vol. 23, pp. 509-520, 2018. http://dx.doi.org/10.1007/s00775-018-1548-6
- J. Montoya, M.J. López-Pérez, and E. Ruiz-Pesini, "Mitochondrial DNA transcription and diseases: Past, present and future", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1757, pp. 1179-1189, 2006. http://dx.doi.org/10.1016/j.bbabio.2006.03.023
- L.A. Zinovkina, "Mechanisms of Mitochondrial DNA Repair in Mammals", Biochemistry (Moscow), vol. 83, pp. 233-249, 2018. http://dx.doi.org/10.1134/S0006297918030045
- E. Ruiz-Pesini, D. Mishmar, M. Brandon, V. Procaccio, and D.C. Wallace, "Effects of Purifying and Adaptive Selection on Regional Variation in Human mtDNA", Science, vol. 303, pp. 223-226, 2004. http://dx.doi.org/10.1126/science.1088434
- J. Montoya, E. López-Gallardo, M.D. Herrero-Martín, �. Martínez-Romero, A. Gómez-Durán, D. Pacheu, M. Carreras, C. Díez-Sánchez, M.J. López-Pérez, and E. Ruiz-Pesini, "Diseases of the Human Mitochondrial Oxidative Phosphorylation System", Advances in Experimental Medicine and Biology, pp. 47-67, 2009. http://dx.doi.org/10.1007/978-90-481-2813-6_5
- A. Kral, and G.M. O'Donoghue, "Profound Deafness in Childhood", New England Journal of Medicine, vol. 363, pp. 1438-1450, 2010. http://dx.doi.org/10.1056/NEJMra0911225
- H. Mutai, T. Watabe, K. Kosaki, K. Ogawa, and T. Matsunaga, "Mitochondrial mutations in maternally inherited hearing loss", BMC Medical Genetics, vol. 18, 2017. http://dx.doi.org/10.1186/s12881-017-0389-4
- R.D.S. Pitceathly, "COX10Mutations Resulting in Complex Multisystem Mitochondrial Disease That Remains Stable Into Adulthood", JAMA Neurology, 2013. http://dx.doi.org/10.1001/jamaneurol.2013.3242
- H. Antonicka, "Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency", Human Molecular Genetics, vol. 12, pp. 2693-2702, 2003. http://dx.doi.org/10.1093/hmg/ddg284
- J.T. Hinson, V.R. Fantin, J. Schönberger, N. Breivik, G. Siem, B. McDonough, P. Sharma, I. Keogh, R. Godinho, F. Santos, A. Esparza, Y. Nicolau, E. Selvaag, B.H. Cohen, C.L. Hoppel, L. Tranebjærg, R.D. Eavey, J. Seidman, and C.E. Seidman, "Missense Mutations in theBCS1LGene as a Cause of the Björnstad Syndrome", New England Journal of Medicine, vol. 356, pp. 809-819, 2007. http://dx.doi.org/10.1056/NEJMoa055262
- E. Fernandez-Vizarra, M. Bugiani, P. Goffrini, F. Carrara, L. Farina, E. Procopio, A. Donati, G. Uziel, I. Ferrero, and M. Zeviani, "Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy", Human Molecular Genetics, vol. 16, pp. 1241-1252, 2007. http://dx.doi.org/10.1093/hmg/ddm072
- M.X. Guan, N. Fischel-Ghodsian, and G. Attardi, "A biochemical basis for the inherited susceptibility to aminoglycoside ototoxicity.", Human molecular genetics, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10915767
-
E. Iglesias, L. Llobet, D. Pacheu‐Grau, A. Gómez‐Durán, and E. Ruiz‐Pesini, "Cybrids for Mitochondrial
DNA Pharmacogenomics", Drug Development Research, vol. 73, pp. 453-460, 2012. http://dx.doi.org/10.1002/ddr.21037 - C. Giordano, F. Pallotti, W.F. Walker, N. Checcarelli, O. Musumeci, F. Santorelli, G. d'Amati, E.A. Schon, S. DiMauro, M. Hirano, and M.M. Davidson, "Pathogenesis of the deafness-associated A1555G mitochondrial DNA mutation", Biochemical and Biophysical Research Communications, vol. 293, pp. 521-529, 2002. http://dx.doi.org/10.1016/S0006-291X(02)00256-5
- D. Pacheu-Grau, A. Gómez-Durán, M.J. López-Pérez, J. Montoya, and E. Ruiz-Pesini, "Mitochondrial pharmacogenomics: barcode for antibiotic therapy", Drug Discovery Today, vol. 15, pp. 33-39, 2010. http://dx.doi.org/10.1016/j.drudis.2009.10.008
- S. Emperador, D. Pacheu-Grau, M.P. Bayona-Bafaluy, N. Garrido-Pérez, A. MartÃn-Navarro, M.J. López-Pérez, J. Montoya, and E. Ruiz-Pesini, "An MRPS12 mutation modifies aminoglycoside sensitivity caused by 12S rRNA mutations", Frontiers in Genetics, vol. 5, 2015. http://dx.doi.org/10.3389/fgene.2014.00469
- D. Pacheu-Grau, A. Gómez-Durán, E. López-Gallardo, T. Pinós, A.L. Andreu, M.J. López-Pérez, J. Montoya, and E. Ruiz-Pesini, "‘Progress’ renders detrimental an ancient mitochondrial DNA genetic variant", Human Molecular Genetics, vol. 20, pp. 4224-4231, 2011. http://dx.doi.org/10.1093/hmg/ddr350
- N. Raimundo, L. Song, T. Shutt, S. McKay, J. Cotney, M. Guan, T. Gilliland, D. Hohuan, J. Santos-Sacchi, and G. Shadel, "Mitochondrial Stress Engages E2F1 Apoptotic Signaling to Cause Deafness", Cell, vol. 148, pp. 716-726, 2012. http://dx.doi.org/10.1016/j.cell.2011.12.027
- J. Finsterer, S. Zarrouk-Mahjoub, and A. Daruich, "The Eye on Mitochondrial Disorders", Journal of Child Neurology, vol. 31, pp. 652-662, 2015. http://dx.doi.org/10.1177/0883073815599263
- Y. Zhu, X. Gu, and C. Xu, "A Mitochondrial DNA A8701G Mutation Partly Associated with Maternally Inherited Hypertension and Dilated Cardiomyopathy in a Chinese Pedigree", Chinese Medical Journal, vol. 129, pp. 1890, 2016. http://dx.doi.org/10.4103/0366-6999.186656
- S. Leruez, P. Amati-Bonneau, C. Verny, P. Reynier, V. Procaccio, D. Bonneau, and D. Milea, "Mitochondrial dysfunction affecting visual pathways", Revue Neurologique, vol. 170, pp. 344-354, 2014. http://dx.doi.org/10.1016/j.neurol.2014.03.009
- J. Finsterer, M. Mancuso, D. Pareyson, J. Burgunder, and T. Klopstock, "Mitochondrial disorders of the retinal ganglion cells and the optic nerve", Mitochondrion, vol. 42, pp. 1-10, 2018. http://dx.doi.org/10.1016/j.mito.2017.10.003
- C. Giordano, L. Iommarini, L. Giordano, A. Maresca, A. Pisano, M.L. Valentino, L. Caporali, R. Liguori, S. Deceglie, M. Roberti, F. Fanelli, F. Fracasso, F.N. Ross-Cisneros, P. D’Adamo, G. Hudson, A. Pyle, P. Yu-Wai-Man, P.F. Chinnery, M. Zeviani, S.R. Salomao, A. Berezovsky, R. Belfort, D.F. Ventura, M. Moraes, M. Moraes Filho, P. Barboni, F. Sadun, A. De Negri, A.A. Sadun, A. Tancredi, M. Mancini, G. d’Amati, P. Loguercio Polosa, P. Cantatore, and V. Carelli, "Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy", Brain, vol. 137, pp. 335-353, 2013. http://dx.doi.org/10.1093/brain/awt343
- C. Giordano, M. Montopoli, E. Perli, M. Orlandi, M. Fantin, F.N. Ross-Cisneros, L. Caparrotta, A. Martinuzzi, E. Ragazzi, A. Ghelli, A.A. Sadun, G. d’Amati, and V. Carelli, "Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy", Brain, vol. 134, pp. 220-234, 2010. http://dx.doi.org/10.1093/brain/awq276
- B.J. Katz, Y. Zhao, J.E. Warner, Z. Tong, Z. Yang, and K. Zhang, "A family with X‐linked optic atrophy linked to the OPA2 locus Xp11.4‐Xp11.2", American Journal of Medical Genetics Part A, vol. 140A, pp. 2207-2211, 2006. http://dx.doi.org/10.1002/ajmg.a.31455
- P.I. Sergouniotis, R. Perveen, D.L. Thiselton, K. Giannopoulos, M. Sarros, J.R. Davies, S. Biswas, A.M. Ansons, J.L. Ashworth, I.C. Lloyd, G.C. Black, and M. Votruba, "Clinical and molecular genetic findings in autosomal dominant OPA3-related optic neuropathy", neurogenetics, vol. 16, pp. 69-75, 2014. http://dx.doi.org/10.1007/s10048-014-0416-y
- J.B. Kerrison, V.J. Arnould, J.M. Ferraz Sallum, M.R. Vagefi, M.M. Barmada, Y. Li, D. Zhu, and I.H. Maumenee, "Genetic heterogeneity of dominant optic atrophy, Kjer type: Identification of a second locus on chromosome 18q12.2-12.3.", Archives of ophthalmology (Chicago, Ill. : 1960), 1999. http://www.ncbi.nlm.nih.gov/pubmed/10369594
- F. Barbet, "A third locus for dominant optic atrophy on chromosome 22q", Journal of Medical Genetics, vol. 42, pp. e1-e1, 2005. http://dx.doi.org/10.1136/jmg.2004.025502
- V. Carelli, S. Schimpf, N. Fuhrmann, M.L. Valentino, C. Zanna, L. Iommarini, M. Papke, S. Schaich, S. Tippmann, B. Baumann, P. Barboni, L. Longanesi, M. Rugolo, A. Ghelli, M.V. Alavi, R.J. Youle, L. Bucchi, R. Carroccia, M.P. Giannoccaro, C. Tonon, R. Lodi, G. Cenacchi, P. Montagna, R. Liguori, and B. Wissinger, "A clinically complex form of dominant optic atrophy (OPA8) maps on chromosome 16", Human Molecular Genetics, vol. 20, pp. 1893-1905, 2011. http://dx.doi.org/10.1093/hmg/ddr071
- H. Eiberg, "Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene", Journal of Medical Genetics, vol. 43, pp. 435-440, 2005. http://dx.doi.org/10.1136/jmg.2005.034892
- R. Anand, T. Wai, M.J. Baker, N. Kladt, A.C. Schauss, E. Rugarli, and T. Langer, "The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission", Journal of Cell Biology, vol. 204, pp. 919-929, 2014. http://dx.doi.org/10.1083/jcb.201308006
- M.J. Baker, P.A. Lampe, D. Stojanovski, A. Korwitz, R. Anand, T. Tatsuta, and T. Langer, "Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics", The EMBO Journal, vol. 33, pp. 578-593, 2014. http://dx.doi.org/10.1002/embj.201386474
- S. Ehses, I. Raschke, G. Mancuso, A. Bernacchia, S. Geimer, D. Tondera, J. Martinou, B. Westermann, E.I. Rugarli, and T. Langer, "Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1", Journal of Cell Biology, vol. 187, pp. 1023-1036, 2009. http://dx.doi.org/10.1083/jcb.200906084
- L. Pernas, and L. Scorrano, "Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function", Annual Review of Physiology, vol. 78, pp. 505-531, 2016. http://dx.doi.org/10.1146/annurev-physiol-021115-105011
- S. Kao, M. Yen, A. Wang, Y. Yeh, and A. Lin, "Changes in Mitochondrial Morphology and Bioenergetics in Human Lymphoblastoid Cells With Four NovelOPA1Mutations", Investigative Opthalmology & Visual Science, vol. 56, pp. 2269, 2015. http://dx.doi.org/10.1167/iovs.14-16288
- T. Wai, J. García-Prieto, M.J. Baker, C. Merkwirth, P. Benit, P. Rustin, F.J. Rupérez, C. Barbas, B. Ibañez, and T. Langer, "Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice", Science, vol. 350, 2015. http://dx.doi.org/10.1126/science.aad0116
- M.D. Metodiev, S. Gerber, L. Hubert, A. Delahodde, D. Chretien, X. Gérard, P. Amati-Bonneau, M. Giacomotto, N. Boddaert, A. Kaminska, I. Desguerre, J. Amiel, M. Rio, J. Kaplan, A. Munnich, A. Rötig, J.M. Rozet, and C. Besmond, "Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy", Journal of Medical Genetics, vol. 51, pp. 834-838, 2014. http://dx.doi.org/10.1136/jmedgenet-2014-102532
- S. Hanein, I. Perrault, O. Roche, S. Gerber, N. Khadom, M. Rio, N. Boddaert, M. Jean-Pierre, N. Brahimi, V. Serre, D. Chretien, N. Delphin, L. Fares-Taie, S. Lachheb, A. Rotig, F. Meire, A. Munnich, J. Dufier, J. Kaplan, and J. Rozet, "TMEM126A, Encoding a Mitochondrial Protein, Is Mutated in Autosomal-Recessive Nonsyndromic Optic Atrophy", The American Journal of Human Genetics, vol. 84, pp. 493-498, 2009. http://dx.doi.org/10.1016/j.ajhg.2009.03.003
- J. Désir, F. Coppieters, N. Van Regemorter, E. De Baere, M. Abramowicz, and M. Cordonnier, "TMEM126A mutation in a Moroccan family with autosomal recessive optic atrophy.", Molecular vision, 2012. http://www.ncbi.nlm.nih.gov/pubmed/22815638
- E. Meyer, M. Michaelides, L.J. Tee, A.G. Robson, F. Rahman, S. Pasha, L.M. Luxon, A.T. Moore, and E.R. Maher, "Nonsense mutation in TMEM126A causing autosomal recessive optic atrophy and auditory neuropathy.", Molecular vision, 2010. http://www.ncbi.nlm.nih.gov/pubmed/20405026
- F. Scaglia, J.A. Towbin, W.J. Craigen, J.W. Belmont, E.O. Smith, S.R. Neish, S.M. Ware, J.V. Hunter, S.D. Fernbach, G.D. Vladutiu, L.C. Wong, and H. Vogel, "Clinical Spectrum, Morbidity, and Mortality in 113 Pediatric Patients With Mitochondrial Disease", Pediatrics, vol. 114, pp. 925-931, 2004. http://dx.doi.org/10.1542/peds.2004-0718
- D. Holmgren, "Cardiomyopathy in children with mitochondrial disease Clinical course and cardiological findings", European Heart Journal, vol. 24, pp. 280-288, 2003. http://dx.doi.org/10.1016/s0195-668x(02)00387-1
- A.W. El-Hattab, and F. Scaglia, "Mitochondrial Cardiomyopathies", Frontiers in Cardiovascular Medicine, vol. 3, 2016. http://dx.doi.org/10.3389/fcvm.2016.00025
- J. Finsterer, and S. Kothari, "Cardiac manifestations of primary mitochondrial disorders", International Journal of Cardiology, vol. 177, pp. 754-763, 2014. http://dx.doi.org/10.1016/j.ijcard.2014.11.014
- R.J. Rodenburg, "Mitochondrial complex I-linked disease", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1857, pp. 938-945, 2016. http://dx.doi.org/10.1016/j.bbabio.2016.02.012
- C. Brunel-Guitton, A. Levtova, and F. Sasarman, "Mitochondrial Diseases and Cardiomyopathies", Canadian Journal of Cardiology, vol. 31, pp. 1360-1376, 2015. http://dx.doi.org/10.1016/j.cjca.2015.08.017
- C.L. Alston, C. Ceccatelli Berti, E.L. Blakely, M. Oláhová, L. He, C.J. McMahon, S.E. Olpin, I.P. Hargreaves, C. Nolli, R. McFarland, P. Goffrini, M.J. O’Sullivan, and R.W. Taylor, "A recessive homozygous p.Asp92Gly SDHD mutation causes prenatal cardiomyopathy and a severe mitochondrial complex II deficiency", Human Genetics, vol. 134, pp. 869-879, 2015. http://dx.doi.org/10.1007/s00439-015-1568-z
- A.L. Andreu, N. Checcarelli, S. Iwata, S. Shanske, and S. Dimauro, "A Missense Mutation in the Mitochondrial Cytochrome b Gene in a Revisited Case with Histiocytoid Cardiomyopathy", Pediatric Research, vol. 48, pp. 311-314, 2000. http://dx.doi.org/10.1203/00006450-200009000-00008
- V. Carossa, A. Ghelli, C.V. Tropeano, M.L. Valentino, L. Iommarini, A. Maresca, L. Caporali, C. La Morgia, R. Liguori, P. Barboni, M. Carbonelli, G. Rizzo, C. Tonon, R. Lodi, A. Martinuzzi, V. De Nardo, M. Rugolo, L. Ferretti, F. Gandini, M. Pala, A. Achilli, A. Olivieri, A. Torroni, and V. Carelli, "A Novel in-Frame 18-bp Microdeletion inMT-CYBCauses a Multisystem Disorder with Prominent Exercise Intolerance", Human Mutation, vol. 35, pp. 954-958, 2014. http://dx.doi.org/10.1002/humu.22596
- H. Antonicka, A. Mattman, C.G. Carlson, D.M. Glerum, K.C. Hoffbuhr, S.C. Leary, N.G. Kennaway, and E.A. Shoubridge, "Mutations in COX15 Produce a Defect in the Mitochondrial Heme Biosynthetic Pathway, Causing Early-Onset Fatal Hypertrophic Cardiomyopathy", The American Journal of Human Genetics, vol. 72, pp. 101-114, 2003. http://dx.doi.org/10.1086/345489
- U.N. Abdulhag, D. Soiferman, O. Schueler-Furman, C. Miller, A. Shaag, O. Elpeleg, S. Edvardson, and A. Saada, "Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy", European Journal of Human Genetics, vol. 23, pp. 159-164, 2014. http://dx.doi.org/10.1038/ejhg.2014.85
- V. Massa, E. Fernandez-Vizarra, S. Alshahwan, E. Bakhsh, P. Goffrini, I. Ferrero, P. Mereghetti, P. D'Adamo, P. Gasparini, and M. Zeviani, "Severe Infantile Encephalomyopathy Caused by a Mutation in COX6B1, a Nucleus-Encoded Subunit of Cytochrome C Oxidase", The American Journal of Human Genetics, vol. 82, pp. 1281-1289, 2008. http://dx.doi.org/10.1016/j.ajhg.2008.05.002
- A. Ghosh, A.T. Pratt, S. Soma, S.G. Theriault, A.T. Griffin, P.P. Trivedi, and V.M. Gohil, "Mitochondrial disease genesCOA6,COX6BandSCO2have overlapping roles in COX2 biogenesis", Human Molecular Genetics, vol. 25, pp. 660-671, 2015. http://dx.doi.org/10.1093/hmg/ddv503
- M. Jaksch, R. Horvath, N. Horn, D.P. Auer, C. Macmillan, J. Peters, K.D. Gerbitz, I. Kraegeloh-Mann, A. Muntau, V. Karcagi, R. Kalmanchey, H. Lochmuller, E.A. Shoubridge, and P. Freisinger, "Homozygosity (E140K) in SCO2 causes delayed infantile onset of cardiomyopathy and neuropathy.", Neurology, 2001. http://www.ncbi.nlm.nih.gov/pubmed/11673586
- B. Mobley, G. Enns, L. Wong, and H. Vogel, "A novel homozygous SCO2 mutation, p.G193S, causing fatal infantile cardioencephalomyopathy", Clinical Neuropathology, vol. 28, pp. 143-149, 2009. http://dx.doi.org/10.5414/npp28143
- L.C. Papadopoulou, C.M. Sue, M.M. Davidson, K. Tanji, I. Nishino, J.E. Sadlock, S. Krishna, W. Walker, J. Selby, D.M. Glerum, R.V. Coster, G. Lyon, E. Scalais, R. Lebel, P. Kaplan, S. Shanske, D.C. De Vivo, E. Bonilla, M. Hirano, S. DiMauro, and E.A. Schon, "Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene", Nature Genetics, vol. 23, pp. 333-337, 1999. http://dx.doi.org/10.1038/15513
- S.C. Leary, H. Antonicka, F. Sasarman, W. Weraarpachai, P.A. Cobine, M. Pan, G.K. Brown, R. Brown, J. Majewski, K.C.H. Ha, S. Rahman, and E.A. Shoubridge, "Novel Mutations inSCO1as a Cause of Fatal Infantile Encephalopathy and Lactic Acidosis", Human Mutation, vol. 34, pp. 1366-1370, 2013. http://dx.doi.org/10.1002/humu.22385
- I. Valnot, S. Osmond, N. Gigarel, B. Mehaye, J. Amiel, V. Cormier-Daire, A. Munnich, J. Bonnefont, P. Rustin, and A. Rötig, "Mutations of the SCO1 Gene in Mitochondrial Cytochrome c Oxidase Deficiency with Neonatal-Onset Hepatic Failure and Encephalopathy", The American Journal of Human Genetics, vol. 67, pp. 1104-1109, 2000. http://dx.doi.org/10.1016/S0002-9297(07)62940-1
- L. Stiburek, K. Vesela, H. Hansikova, H. Hulkova, and J. Zeman, "Loss of function of Sco1 and its interaction with cytochrome c oxidase", American Journal of Physiology-Cell Physiology, vol. 296, pp. C1218-C1226, 2009. http://dx.doi.org/10.1152/ajpcell.00564.2008
- S.C. Leary, "Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase", Human Molecular Genetics, vol. 13, pp. 1839-1848, 2004. http://dx.doi.org/10.1093/hmg/ddh197
- S.C. Leary, P.A. Cobine, B.A. Kaufman, G. Guercin, A. Mattman, J. Palaty, G. Lockitch, D.R. Winge, P. Rustin, R. Horvath, and E.A. Shoubridge, "The Human Cytochrome c Oxidase Assembly Factors SCO1 and SCO2 Have Regulatory Roles in the Maintenance of Cellular Copper Homeostasis", Cell Metabolism, vol. 5, pp. 9-20, 2007. http://dx.doi.org/10.1016/j.cmet.2006.12.001
- Z.N. Baker, K. Jett, A. Boulet, A. Hossain, P.A. Cobine, B. Kim, A.M. El Zawily, L. Lee, G.F. Tibbits, M.J. Petris, and S.C. Leary, "The mitochondrial metallochaperone SCO1 maintains CTR1 at the plasma membrane to preserve copper homeostasis in the murine heart", Human Molecular Genetics, vol. 26, pp. 4617-4628, 2017. http://dx.doi.org/10.1093/hmg/ddx344
- S.E. Calvo, A.G. Compton, S.G. Hershman, S.C. Lim, D.S. Lieber, E.J. Tucker, A. Laskowski, C. Garone, S. Liu, D.B. Jaffe, J. Christodoulou, J.M. Fletcher, D.L. Bruno, J. Goldblatt, S. DiMauro, D.R. Thorburn, and V.K. Mootha, "Molecular Diagnosis of Infantile Mitochondrial Disease with Targeted Next-Generation Sequencing", Science Translational Medicine, vol. 4, 2012. http://dx.doi.org/10.1126/scitranslmed.3003310
- F. Baertling, M. A.M. van den Brand, J.L. Hertecant, A. Al-Shamsi, L. P. van den Heuvel, F. Distelmaier, E. Mayatepek, J.A. Smeitink, L.G. Nijtmans, and R.J. Rodenburg, "Mutations inCOA6cause CytochromecOxidase Deficiency and Neonatal Hypertrophic Cardiomyopathy", Human Mutation, vol. 36, pp. 34-38, 2014. http://dx.doi.org/10.1002/humu.22715
- A. Ghosh, P.P. Trivedi, S.A. Timbalia, A.T. Griffin, J.J. Rahn, S.S.L. Chan, and V.M. Gohil, "Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of COA6 deficiency", Human Molecular Genetics, vol. 23, pp. 3596-3606, 2014. http://dx.doi.org/10.1093/hmg/ddu069
- F. Vögtle, J.M. Burkhart, S. Rao, C. Gerbeth, J. Hinrichs, J. Martinou, A. Chacinska, A. Sickmann, R.P. Zahedi, and C. Meisinger, "Intermembrane Space Proteome of Yeast Mitochondria", Molecular & Cellular Proteomics, vol. 11, pp. 1840-1852, 2012. http://dx.doi.org/10.1074/mcp.M112.021105
- D. Pacheu-Grau, B. Bareth, J. Dudek, L. Juris, F. Vögtle, M. Wissel, S. Leary, S. Dennerlein, P. Rehling, and M. Deckers, "Cooperation between COA6 and SCO2 in COX2 Maturation during Cytochrome c Oxidase Assembly Links Two Mitochondrial Cardiomyopathies", Cell Metabolism, vol. 21, pp. 823-833, 2015. http://dx.doi.org/10.1016/j.cmet.2015.04.012
- D.A. Stroud, M.J. Maher, C. Lindau, F. Vögtle, A.E. Frazier, E. Surgenor, H. Mountford, A.P. Singh, M. Bonas, S. Oeljeklaus, B. Warscheid, C. Meisinger, D.R. Thorburn, and M.T. Ryan, "COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2", Human Molecular Genetics, vol. 24, pp. 5404-5415, 2015. http://dx.doi.org/10.1093/hmg/ddv265
- M. Bourens, and A. Barrientos, "Human mitochondrial cytochrome c oxidase assembly factor COX18 acts transiently as a membrane insertase within the subunit 2 maturation module", Journal of Biological Chemistry, vol. 292, pp. 7774-7783, 2017. http://dx.doi.org/10.1074/jbc.M117.778514
- Z. Liu, Y. Song, D. Li, X. He, S. Li, B. Wu, W. Wang, S. Gu, X. Zhu, X. Wang, Q. Zhou, Y. Dai, and Q. Yan, "The novel mitochondrial 16S rRNA 2336T>C mutation is associated with hypertrophic cardiomyopathy", Journal of Medical Genetics, vol. 51, pp. 176-184, 2013. http://dx.doi.org/10.1136/jmedgenet-2013-101818
- F.M. Santorelli, K. Tanji, P. Manta, C. Casali, S. Krishna, A.P. Hays, D.M. Mancini, S. DiMauro, and M. Hirano, "Maternally Inherited Cardiomyopathy: An Atypical Presentation of the mtDNA 12S rRNA Gene A1555G Mutation", The American Journal of Human Genetics, vol. 64, pp. 295-300, 1999. http://dx.doi.org/10.1086/302188
- S. Ahola, P. Isohanni, L. Euro, V. Brilhante, A. Palotie, H. Pihko, T. Lönnqvist, T. Lehtonen, J. Laine, H. Tyynismaa, and A. Suomalainen, "Mitochondrial EFTs defects in juvenile-onset Leigh disease, ataxia, neuropathy, and optic atrophy", Neurology, vol. 83, pp. 743-751, 2014. http://dx.doi.org/10.1212/WNL.0000000000000716
- F. Distelmaier, T.B. Haack, C.B. Catarino, C. Gallenmüller, R.J. Rodenburg, T.M. Strom, F. Baertling, T. Meitinger, E. Mayatepek, H. Prokisch, and T. Klopstock, "MRPL44 mutations cause a slowly progressive multisystem disease with childhood-onset hypertrophic cardiomyopathy", neurogenetics, vol. 16, pp. 319-323, 2015. http://dx.doi.org/10.1007/s10048-015-0444-2
- L. Galmiche, V. Serre, M. Beinat, Z. Assouline, A. Lebre, D. Chretien, P. Nietschke, V. Benes, N. Boddaert, D. Sidi, F. Brunelle, M. Rio, A. Munnich, and A. Rötig, "Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy", Human Mutation, vol. 32, pp. 1225-1231, 2011. http://dx.doi.org/10.1002/humu.21562
- S. Emperador, M.P. Bayona-Bafaluy, A. Fernández-Marmiesse, M. Pineda, B. Felgueroso, E. López-Gallardo, R. Artuch, I. Roca, E. Ruiz-Pesini, M.L. Couce, and J. Montoya, "Molecular-genetic characterization and rescue of a TSFM mutation causing childhood-onset ataxia and nonobstructive cardiomyopathy", European Journal of Human Genetics, vol. 25, pp. 153-156, 2016. http://dx.doi.org/10.1038/ejhg.2016.124
- C. Giordano, E. Perli, M. Orlandi, A. Pisano, H.A. Tuppen, L. He, R. Ierinò, L. Petruzziello, A. Terzi, C. Autore, V. Petrozza, P. Gallo, R.W. Taylor, and G. d'Amati, "Cardiomyopathies due to homoplasmic mitochondrial tRNA mutations: morphologic and molecular features", Human Pathology, vol. 44, pp. 1262-1270, 2013. http://dx.doi.org/10.1016/j.humpath.2012.10.011
- J.D. Goldstein, S. Shanske, C. Bruno, and A.A. Perszyk, "Maternally Inherited Mitochondrial Cardiomyopathy Associated with a C-to-T Transition at Nucleotide 3303 of Mitochondrial DNA in the tRNALeu(UUR) Gene", Pediatric and Developmental Pathology, vol. 2, pp. 78-85, 1999. http://dx.doi.org/10.1007/s100249900094
- O. Alila-Fersi, M. Tabebi, M. Maalej, N. Belguith, L. Keskes, E. Mkaouar-Rebai, and F. Fakhfakh, "First description of a novel mitochondrial mutation in the MT-TI gene associated with multiple mitochondrial DNA deletion and depletion in family with severe dilated mitochondrial cardiomyopathy", Biochemical and Biophysical Research Communications, vol. 497, pp. 1049-1054, 2018. http://dx.doi.org/10.1016/j.bbrc.2018.02.173
- P.G. Barth, F. Valianpour, V.M. Bowen, J. Lam, M. Duran, F.M. Vaz, and R.J. Wanders, "X‐linked cardioskeletal myopathy and neutropenia (Barth syndrome): An update", American Journal of Medical Genetics Part A, vol. 126A, pp. 349-354, 2004. http://dx.doi.org/10.1002/ajmg.a.20660
- P.G. Barth, C. Van den Bogert, P.A. Bolhuis, H.R. Scholte, A.H. van Gennip, R.B.H. Schutgens, and A.G. Ketel, "X‐linked cardioskeletal myopathy and neutropenia (Barth syndrome): Respiratory‐chain abnormalities in cultured fibroblasts", Journal of Inherited Metabolic Disease, vol. 19, pp. 157-160, 1996. http://dx.doi.org/10.1007/bf01799418
- J. Dudek, and C. Maack, "Barth syndrome cardiomyopathy", Cardiovascular Research, vol. 113, pp. 399-410, 2017. http://dx.doi.org/10.1093/cvr/cvx014
- Y. Xu, A. Malhotra, M. Ren, and M. Schlame, "The Enzymatic Function of Tafazzin", Journal of Biological Chemistry, vol. 281, pp. 39217-39224, 2006. http://dx.doi.org/10.1074/jbc.M606100200
- M. Vukotic, H. Nolte, T. König, S. Saita, M. Ananjew, M. Krüger, T. Tatsuta, and T. Langer, "Acylglycerol Kinase Mutated in Sengers Syndrome Is a Subunit of the TIM22 Protein Translocase in Mitochondria", Molecular Cell, vol. 67, pp. 471-483.e7, 2017. http://dx.doi.org/10.1016/j.molcel.2017.06.013
- Y. Kang, D.A. Stroud, M.J. Baker, D.P. De Souza, A.E. Frazier, M. Liem, D. Tull, S. Mathivanan, M.J. McConville, D.R. Thorburn, M.T. Ryan, and D. Stojanovski, "Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex", Molecular Cell, vol. 67, pp. 457-470.e5, 2017. http://dx.doi.org/10.1016/j.molcel.2017.06.014
- C.T. Moraes, S. DiMauro, M. Zeviani, A. Lombes, S. Shanske, A.F. Miranda, H. Nakase, E. Bonilla, L.C. Werneck, S. Servidei, I. Nonaka, Y. Koga, A.J. Spiro, A.K. W. Brownell, B. Schmidt, D.L. Schotland, M. Zupanc, D.C. DeVivo, E.A. Schon, and L.P. Rowland, "Mitochondrial DNA Deletions in Progressive External Ophthalmoplegia and Kearns-Sayre Syndrome", New England Journal of Medicine, vol. 320, pp. 1293-1299, 1989. http://dx.doi.org/10.1056/NEJM198905183202001
- D.M. Kirby, "Mutations of the mitochondrial ND1 gene as a cause of MELAS", Journal of Medical Genetics, vol. 41, pp. 784-789, 2004. http://dx.doi.org/10.1136/jmg.2004.020537
- F.M. Santorelli, K. Tanji, R. Kulikova, S. Shanske, L. Vilarinho, A.P. Hays, and S. DiMauro, "Identification of a Novel Mutation in the mtDNA ND5 Gene Associated with MELAS", Biochemical and Biophysical Research Communications, vol. 238, pp. 326-328, 1997. http://dx.doi.org/10.1006/bbrc.1997.7167
- Y. Goto, I. Nonaka, and S. Horai, "A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies", Nature, vol. 348, pp. 651-653, 1990. http://dx.doi.org/10.1038/348651a0
- G. Manfredi, E. Schon, C. Moraes, E. Bonilla, G. Berry, J. Sladky, and S. Dimauro, "A new mutation associated with MELAS is located in a mitochondrial DNA polypeptide-coding gene", Neuromuscular Disorders, vol. 5, pp. 391-398, 1995. http://dx.doi.org/10.1016/0960-8966(94)00079-o
- D.C. Wallace, X. Zheng, M.T. Lott, J.M. Shoffner, J.A. Hodge, R.I. Kelley, C.M. Epstein, and L.C. Hopkins, "Familial mitochondrial encephalomyopathy (MERRF): Genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease", Cell, vol. 55, pp. 601-610, 1988. http://dx.doi.org/10.1016/0092-8674(88)90218-8
- M. Zeviani, F. Muntoni, N. Savarese, G. Serra, V. Tiranti, F. Carrara, C. Mariotti, and S. DiDonato, "A MERRF/MELAS Overlap Syndrome Associated with a New Point Mutation in the Mitochondrial DNA tRNA^Lys Gene", European Journal of Human Genetics, vol. 1, pp. 80-87, 1993. http://dx.doi.org/10.1159/000472390
- M. Ozawa, I. Nishino, S. Horai, I. Nonaka, and Y.I. Goto, "Myoclonus epilepsy associated with ragged-red fibers: a G-to-A mutation at nucleotide pair 8363 in mitochondrial tRNA(Lys) in two families.", Muscle & nerve, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9052804
- "Myoclonic epilepsy and ragged red fiber (MERRF) disease", Atlas of Inherited Metabolic Diseases 3E, pp. 382-387, 2011. http://dx.doi.org/10.1201/b15310-58
-
N. Nissanka, and C.T. Moraes, "Mitochondrial
DNA damage and reactive oxygen species in neurodegenerative disease", FEBS Letters, vol. 592, pp. 728-742, 2018. http://dx.doi.org/10.1002/1873-3468.12956 - I.J. Holt, A.E. Harding, R.K. Petty, and J.A. Morgan-Hughes, "A new mitochondrial disease associated with mitochondrial DNA heteroplasmy.", American journal of human genetics, 1990. http://www.ncbi.nlm.nih.gov/pubmed/2137962
- N.J. Lake, A.G. Compton, S. Rahman, and D.R. Thorburn, "Leigh syndrome: One disorder, more than 75 monogenic causes", Annals of Neurology, vol. 79, pp. 190-203, 2015. http://dx.doi.org/10.1002/ana.24551
- A. Wojtala, A. Karkucinska-Wieckowska, V.A. Sardao, J. Szczepanowska, P. Kowalski, M. Pronicki, J. Duszynski, and M.R. Wieckowski, "Modulation of mitochondrial dysfunction-related oxidative stress in fibroblasts of patients with Leigh syndrome by inhibition of prooxidative p66Shc pathway", Mitochondrion, vol. 37, pp. 62-79, 2017. http://dx.doi.org/10.1016/j.mito.2017.07.002
- A. Spinazzola, C. Viscomi, E. Fernandez-Vizarra, F. Carrara, P. D'Adamo, S. Calvo, R.M. Marsano, C. Donnini, H. Weiher, P. Strisciuglio, R. Parini, E. Sarzi, A. Chan, S. DiMauro, A. Rötig, P. Gasparini, I. Ferrero, V.K. Mootha, V. Tiranti, and M. Zeviani, "MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion", Nature Genetics, vol. 38, pp. 570-575, 2006. http://dx.doi.org/10.1038/ng1765
- A. Spinazzola, R. Santer, O.H. Akman, K. Tsiakas, H. Schaefer, X. Ding, C.L. Karadimas, S. Shanske, J. Ganesh, S. Di Mauro, and M. Zeviani, "Hepatocerebral Form of Mitochondrial DNA Depletion Syndrome", Archives of Neurology, vol. 65, 2008. http://dx.doi.org/10.1001/archneur.65.8.1108
- A. Spinazzola, V. Massa, M. Hirano, and M. Zeviani, "Lack of founder effect for an identical mtDNA depletion syndrome (MDS)-associated MPV17 mutation shared by Navajos and Italians", Neuromuscular Disorders, vol. 18, pp. 315-318, 2008. http://dx.doi.org/10.1016/j.nmd.2007.12.007
- A.W. El-Hattab, J. Wang, H. Dai, M. Almannai, C. Staufner, M. Alfadhel, M.J. Gambello, P. Prasun, S. Raza, H.J. Lyons, M. Afqi, M.A.M. Saleh, E.A. Faqeih, H.I. Alzaidan, A. Alshenqiti, L.A. Flore, J. Hertecant, S. Sacharow, D.S. Barbouth, K. Murayama, A.A. Shah, H.C. Lin, and L.C. Wong, "MPV17-related mitochondrial DNA maintenance defect: New cases and review of clinical, biochemical, and molecular aspects", Human Mutation, vol. 39, pp. 461-470, 2018. http://dx.doi.org/10.1002/humu.23387
- N. Miyake, S. Yamashita, K. Kurosawa, S. Miyatake, Y. Tsurusaki, H. Doi, H. Saitsu, and N. Matsumoto, "A novel homozygous mutation of DARS2 may cause a severe LBSL variant", Clinical Genetics, vol. 80, pp. 293-296, 2011. http://dx.doi.org/10.1111/j.1399-0004.2011.01644.x
- S. Şahin, A. Cansu, E. Kalay, T. Dinçer, S. Kul, �.M. Çakır, T. Kamaşak, and G.Y. Budak, "Leukoencephalopathy with thalamus and brainstem involvement and high lactate caused by novel mutations in the EARS2 gene in two siblings", Journal of the Neurological Sciences, vol. 365, pp. 54-58, 2016. http://dx.doi.org/10.1016/j.jns.2016.04.008
-
V. Boczonadi, M.J. Jennings, and R. Horvath, "The role of
tRNA synthetases in neurological and neuromuscular disorders", FEBS Letters, vol. 592, pp. 703-717, 2018. http://dx.doi.org/10.1002/1873-3468.12962 - C. Viscomi, "Toward a therapy for mitochondrial disease", Biochemical Society Transactions, vol. 44, pp. 1483-1490, 2016. http://dx.doi.org/10.1042/BST20160085
- R.C. Scarpulla, "Transcriptional Paradigms in Mammalian Mitochondrial Biogenesis and Function", Physiological Reviews, vol. 88, pp. 611-638, 2008. http://dx.doi.org/10.1152/physrev.00025.2007
- P. Puigserver, and B.M. Spiegelman, "Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α (PGC-1α): Transcriptional Coactivator and Metabolic Regulator", Endocrine Reviews, vol. 24, pp. 78-90, 2003. http://dx.doi.org/10.1210/er.2002-0012
-
J.A. Villena, "New insights into
PGC ‐1 coactivators: redefining their role in the regulation of mitochondrial function and beyond", The FEBS Journal, vol. 282, pp. 647-672, 2015. http://dx.doi.org/10.1111/febs.13175 - E. Perales-Clemente, M.P. Bayona-Bafaluy, A. Pérez-Martos, A. Barrientos, P. Fernández-Silva, and J.A. Enriquez, "Restoration of electron transport without proton pumping in mammalian mitochondria", Proceedings of the National Academy of Sciences, vol. 105, pp. 18735-18739, 2008. http://dx.doi.org/10.1073/pnas.0810518105
- E.P. Dassa, E. Dufour, S. Gonçalves, V. Paupe, G.A.J. Hakkaart, H.T. Jacobs, and P. Rustin, "Expression of the alternative oxidase complements cytochrome c oxidase deficiency in human cells", EMBO Molecular Medicine, vol. 1, pp. 30-36, 2009. http://dx.doi.org/10.1002/emmm.200900001
- K.K. Kemppainen, J. Rinne, A. Sriram, M. Lakanmaa, A. Zeb, T. Tuomela, A. Popplestone, S. Singh, A. Sanz, P. Rustin, and H.T. Jacobs, "Expression of alternative oxidase in Drosophila ameliorates diverse phenotypes due to cytochrome oxidase deficiency", Human Molecular Genetics, vol. 23, pp. 2078-2093, 2013. http://dx.doi.org/10.1093/hmg/ddt601
- A.F. Camargo, M.M. Chioda, A.P.C. Rodrigues, G.S. Garcia, E.A. McKinney, H.T. Jacobs, and M.T. Oliveira, "Xenotopic expression of alternative electron transport enzymes in animal mitochondria and their impact in health and disease", Cell Biology International, vol. 42, pp. 664-669, 2018. http://dx.doi.org/10.1002/cbin.10943
- M. Szibor, P.K. Dhandapani, E. Dufour, K.M. Holmström, Y. Zhuang, I. Salwig, I. Wittig, J. Heidler, Z. Gizatullina, T. Gainutdinov, G.M.C. Consortium, H. Fuchs, V. Gailus-Durner, M.H. de Angelis, J. Nandania, V. Velagapudi, A. Wietelmann, P. Rustin, F.N. Gellerich, H.T. Jacobs, and T. Braun, "Broad AOX expression in a genetically tractable mouse model does not disturb normal physiology", Disease Models & Mechanisms, 2016. http://dx.doi.org/10.1242/dmm.027839
- G. Civiletto, T. Varanita, R. Cerutti, T. Gorletta, S. Barbaro, S. Marchet, C. Lamperti, C. Viscomi, L. Scorrano, and M. Zeviani, "Opa1 Overexpression Ameliorates the Phenotype of Two Mitochondrial Disease Mouse Models", Cell Metabolism, vol. 21, pp. 845-854, 2015. http://dx.doi.org/10.1016/j.cmet.2015.04.016
- T. Varanita, M. Soriano, V. Romanello, T. Zaglia, R. Quintana-Cabrera, M. Semenzato, R. Menabò, V. Costa, G. Civiletto, P. Pesce, C. Viscomi, M. Zeviani, F. Di Lisa, M. Mongillo, M. Sandri, and L. Scorrano, "The Opa1-Dependent Mitochondrial Cristae Remodeling Pathway Controls Atrophic, Apoptotic, and Ischemic Tissue Damage", Cell Metabolism, vol. 21, pp. 834-844, 2015. http://dx.doi.org/10.1016/j.cmet.2015.05.007
- J. Beyrath, M. Pellegrini, H. Renkema, L. Houben, S. Pecheritsyna, P. van Zandvoort, P. van den Broek, A. Bekel, P. Eftekhari, and J.A.M. Smeitink, "KH176 Safeguards Mitochondrial Diseased Cells from Redox Stress-Induced Cell Death by Interacting with the Thioredoxin System/Peroxiredoxin Enzyme Machinery", Scientific Reports, vol. 8, 2018. http://dx.doi.org/10.1038/s41598-018-24900-3
- W.J. Koopman, J. Beyrath, C. Fung, S. Koene, R.J. Rodenburg, P.H. Willems, and J.A. Smeitink, "Mitochondrial disorders in children: toward development of small‐molecule treatment strategies", EMBO Molecular Medicine, vol. 8, pp. 311-327, 2016. http://dx.doi.org/10.15252/emmm.201506131
- S.R. Bacman, S.L. Williams, M. Pinto, S. Peralta, and C.T. Moraes, "Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs", Nature Medicine, vol. 19, pp. 1111-1113, 2013. http://dx.doi.org/10.1038/nm.3261
-
P.A. Gammage, J. Rorbach, A.I. Vincent, E.J. Rebar, and M. Minczuk, "Mitochondrially targeted
ZFN s for selective degradation of pathogenic mitochondrial genomes bearing large‐scale deletions or point mutations", EMBO Molecular Medicine, vol. 6, pp. 458-466, 2014. http://dx.doi.org/10.1002/emmm.201303672 - G. Wang, E. Shimada, J. Zhang, J.S. Hong, G.M. Smith, M.A. Teitell, and C.M. Koehler, "Correcting human mitochondrial mutations with targeted RNA import", Proceedings of the National Academy of Sciences, vol. 109, pp. 4840-4845, 2012. http://dx.doi.org/10.1073/pnas.1116792109
- P.A. Gammage, C.T. Moraes, and M. Minczuk, "Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized", Trends in Genetics, vol. 34, pp. 101-110, 2018. http://dx.doi.org/10.1016/j.tig.2017.11.001
- A.E. Frazier, D.R. Thorburn, and A.G. Compton, "Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology", Journal of Biological Chemistry, vol. 294, pp. 5386-5395, 2019. http://dx.doi.org/10.1074/jbc.R117.809194
- J. Nikkanen, S. Forsström, L. Euro, I. Paetau, R. Kohnz, L. Wang, D. Chilov, J. Viinamäki, A. Roivainen, P. Marjamäki, H. Liljenbäck, S. Ahola, J. Buzkova, M. Terzioglu, N. Khan, S. Pirnes-Karhu, A. Paetau, T. Lönnqvist, A. Sajantila, P. Isohanni, H. Tyynismaa, D. Nomura, B. Battersby, V. Velagapudi, C. Carroll, and A. Suomalainen, "Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism", Cell Metabolism, vol. 23, pp. 635-648, 2016. http://dx.doi.org/10.1016/j.cmet.2016.01.019
- N.A. Khan, J. Nikkanen, S. Yatsuga, C. Jackson, L. Wang, S. Pradhan, R. Kivelä, A. Pessia, V. Velagapudi, and A. Suomalainen, "mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression", Cell Metabolism, vol. 26, pp. 419-428.e5, 2017. http://dx.doi.org/10.1016/j.cmet.2017.07.007
- D. Pacheu-Grau, A. Gomez-Duran, E. Iglesias, E. Lopez-Gallardo, J. Montoya, and E. Ruiz-Pesini, "Mitochondrial antibiograms in personalized medicine", Human Molecular Genetics, vol. 22, pp. 1132-1139, 2012. http://dx.doi.org/10.1093/hmg/dds517
ACKNOWLEDGMENTS
We are indebted to Sylvie Callegari for critical reading of the manuscript and to Peter Rehling and Alexander Schendzielorz for fruitful discussion.
COPYRIGHT
© 2018

Mitochondrial dysfunction and its role in tissue-specific cellular stress by Pacheu-Grau et al. is licensed under a Creative Commons Attribution 4.0 International License.


