Reviews:
Cell Stress, Vol. 3, No. 6, pp. 165 - 180; doi: 10.15698/cst2019.06.188
Biology and clinical relevance of EpCAM

1 Institute of Tumor Biology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
§ These authors contributed equally to the work.
Keywords: EpCAM, liquid biopsy, circulating tumor cells, EMT, Tumor biomarker, tumor cell dissemination.
Abbreviatons:
aa – amino acid,
CD – C-Domain,
CTC – circulating tumor cell,
CTE – congenital tufting enteropathy,
EMT – Epithelial-Mesenchymal Transition,
EpCAM – epithelial cell adhesion molecule,
EV – extracellular vesicle,
KO – kockout,
RIP – Regulated Intramembrane Proteolysis,
TEM – tetraspanin-enriched microdomains,
TM – transmembrane,
TY – Thyroglobulin type 1A domain.
Received originally: 04/02/2019 Received in revised form: 10/04/2019
Accepted: 16/04/2019
Published: 21/05/2019
Correspondence:
Klaus Pantel, Phone: +494074105-3503 pantel@uke.de
Conflict of interest statement: K.P. has ongoing patent applications related to circulat-ing tumor cells. K.P. has received honoraria from Agena, Novartis, Roche and Sanofi and research funding from European Federation of Pharmaceutical Industries and Associations (EFPIA) partners (Angle, Menarini and Ser-vier) of the CANCER-ID programme of the European Un-ion–EFPIA Innovative Medicines Initiative. L.K and S.W. declare no conflict of interest.
Please cite this article as: Laura Keller, Stefan Werner and Klaus Pantel (2019). Biology and clinical relevance of EpCAM. Cell Stress 3(6): 165-180. doi: 10.15698/cst2019.06.188
Abstract
Epithelial cell adhesion molecule (EpCAM) is a transmembrane glycoprotein primarily known to mediate homotypic cell contacts in epithelia tissues. Because EpCAM expression is limited to normal and malignant epithelia, it has been used as diagnostic marker for the detection of carcinoma cells in mesenchymal organs such as blood, bone marrow or lymph nodes. In particular, the detection and molecular characterization of EpCAM-positive circulating tumor cells (CTCs) in the blood of carcinoma patients has gained considerable interest over the past ten years. EpCAM is primarily considered as an adhesion molecule, but recent studies have shown diverse biological functions including regulation of cell proliferation and cancer stemness. In this review, we summarize the current knowledge on the biological properties of EpCAM with emphasis on mechanisms involved in cancer progression and discuss the clinical implications of these findings for the clinical use of EpCAM as a diagnostic marker.
INTRODUCTION
The EpCAM protein was discovered almost 40 years ago as a major epithelial carcinoma antigen by M. Herlyn and colleagues, as a result of its property to generate monoclonal antibodies binding specifically to human colorectal carcinoma cells [1]. In the following, the protein has been independently described many times as a highly immunogenic tumor-associated antigen. In these studies, the discovered antigen received the name of the respective monoclonal antibody recognizing it (a summary table of its different names can be found in [2] and [3]). Specifications of the identified antigens and subsequent cloning of the corresponding genes, in each case, lead to their identity as EpCAM [4]. Since 2007, the nomenclature has been harmonized and it has been agreed that the protein as well as its encoding gene (EPCAM) shall be called EpCAM, which is the abbreviation for Epithelial Cell Adhesion Molecule due to the first reports from Litvinov and colleagues on its adherent function in epithelial cells [4].
–
Liquid biopsy in oncology has gained increasing interest during the last decade revealing potential to change clinical practice by exploiting peripheral blood as a source of information about tumor status and treatment options [5]. Liquid biopsy is a general denomination introduced by Pantel and Alix-Panabieres approximately ten years ago [6], which refers to any tumorderived analyte present in body fluids like peripheral blood, urine, bone marrow and salvia. Nevertheless, of peculiar interest is the detection of circulating tumor cells (CTCs) and tumor-derived soluble molecules or particles, such as circulating tumor DNA (ctDNA) and other circulating nucleic acids (in particular microRNAs), extracellular vesicles and tumor-educated platelets in the blood circulation of cancer patients. The greatest challenge of this field is to specifically discriminate the tumor-derived analyte from a tremendously high background composed of healthy cells and their content. The majority of publications on liquid biopsy focuses on CTCs that allow to obtain a broad range of information at the DNA, RNA and protein level [7][8][9][10]. As the vast majority of cancers are of epithelial origin, targeting epithelial antigens was the first approach to discriminate a tumor cell among millions of peripheral blood mononuclear cells that are of mesenchymal origin. EpCAM therefore became the most commonly used epithelial marker for the capture of CTCs in the blood circulation of carcinoma patients.
–
Here, we will review the accumulating recent evidence that EpCAM is a special tumor marker with profound biological properties far beyond inter-cellular adhesion. This knowledge will open new avenues for the use of EpCAM as a diagnostic liquid biopsy marker in cancer patients.
BIOLOGICAL PROPERTIES OF EpCAM
EPCAM gene mutations
The EPCAM gene consists of 14 kb in total and is located on chromosome 2 (2p21). The gene is conserved across many different species from zebrafish to humans. Particularly the amino acid (aa) sequence of the extracellular domain is conserved to a high extent from fishes to primates, suggesting the functional importance of the EpCAM protein [11]. Mutations in the EPCAM gene have been identified in two hereditary syndromes. In congenital tufting enteropathy (CTE), a rare autosomal recessive form of intractable diarrhea of infancy and Lynch Syndrome also known as Hereditary Non-Polyposis Colorectal Cancer (HNPCC), which is one of the most common cancer susceptibility syndromes that predisposes to colorectal adenocarcinoma, endometrial carcinoma, and various other cancers. In CTE, biallelic EPCAM mutations are mostly loss of functions mutations, predicted to affect EpCAM protein structure, disrupting its expression and/or stability [12]. Constitutive and inducible CTE-associated murine models have been developed by engineering EPCAM KO mice. These models show enhanced intestinal permeability and migration as well as decreased ion transport. The consequences of EpCAM loss in this disease are complex, including decreased expression of tight junctional proteins like Claudins [13][14] or dysregulation of E Cadherin and ß Catenin leading to disorganized transition from crypt to villi [15]. Lynch syndrome is caused by inheritance of one defective allele in genes involved in DNA mismatch repair (MMR) machinery, predominantly MSH2, MLH1, MS2 and MSH6. Contrary to CTE, EPCAM-associated Lynch syndrome is not due to loss of EpCAM per se, but rather is due to monoallelic deletions of the 3′ end of the EPCAM gene in which the polyadenylation signal is lost leading to MSH2 promoter hypermethylation, read-through transcription of the EPCAM and MSH2 genes, and loss of MSH2 protein expression [16].
–
EpCAM protein structure
Human EpCAM protein is a transmembrane glycoprotein polypeptide of 314 aa, consisting of a large N-terminal extracellular domain (EpEX) of 242 aa and 27 kDa, a single-spanning transmembrane domain (TM) of 23 aa and 2 kDa and a short C-terminal cytoplasmic domain of 26 aa and 3 kDa (EpIC; Figure 1).
–
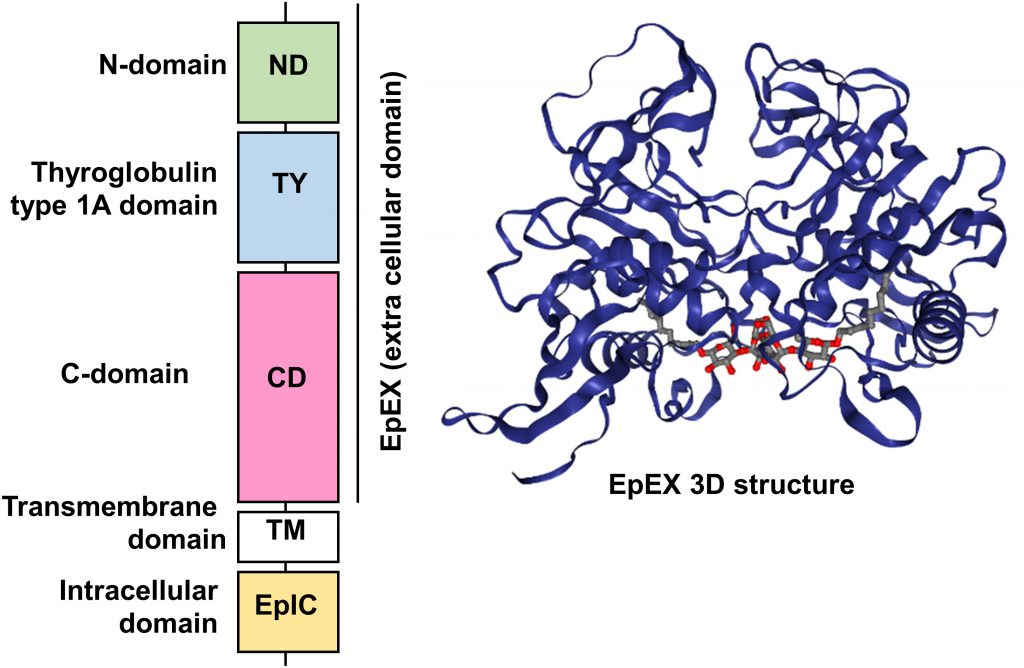 | FIGURE 1: Schematic diagram of the domain structure of full length EpCAM protein and crystal structure of an extracellular EpCAM cis-homodimer according to Pavsic et al. Full length EpCAM consists of a N-terminal signal peptide (SP) followed by three compactly folded extracellular domains (N-Domain (ND), Thyroglobulin type 1A domain (TY) C-Domain CD), a single spanning transmembrane domain (TM) and a c-terminal intracellular domain (EpIC). Two EpCAM subunits form a heart-shaped dimer on the cell surface [11]. Protein Data Base entry 4MZV. |
–
Significant insights in the structure of the EpCAM protein were recently gained thanks to the crystallization of a non-glycosylated form of the EpEX domain by Pavsic and colleagues that is lacking the N-terminal signal peptide [11]. The authors found out that the extracellular part of human EpCAM forms a heart-shaped dimer, which would form at cell surfaces. The polypeptide chain of EpEX is folded into a compact shape made of three domains (N-Domain ND, Thyroglobulin type 1A domain TY, and C-Domain CD) arranged in a triangular fashion where each domain contacts the other two. The extracellular domain also presents three N-glycosylation sites (Asn74, 111, 198), implicated in protein stability and covering the lateral protein surfaces (Figure 1) [11].
–
One function of the intracellular cytoplasmic domain is to anchor the EpCAM protein to the cytoskeleton, as demonstrated by Balzar and colleagues via an interaction with α-actinin [17]. At the C-terminus, amino acids 312-314 display a putative PDZ binding site, which has been shown in other intercellular contact proteins to be key in complex formation with signaling or structural proteins [2]. In line with this, a short segment of the cytoplasmic tail was found to resemble the inhibitory domains of PKCs and could cause PKC inhibition [18]. In vivo, EpCAM forms a cis-dimer of two EpCAM molecules on the surface of the same cell that approximately protrude 5 nm form the cell surface. The dimerization depends on the loop of the TY domain (involved in interactions with the CD of the other molecule) and also on the transmembrane helix [11]. We will discuss in the following chapter how these structural insights can help to refine the different cellular roles of EpCAM.
–
The EpCAM protein contains several cleavage sites that are essential for its biological activity as well as for controlling protein expression. It is worth mentioning that soon after identification of the EpCAM protein, a cleavage at position Arg-80/Arg-81 of the TY loop was discovered. Cleavage of EpCAM at this position results in a 6 kDa N-terminal fragment that remains bound to the protein backbone by the first disulfide bond within the TY-like domain and importantly, would lead to the disruption of the cis-dimer as experimentally demonstrated on the EpEX domain [11]. In vivo, this cleavage is supposed not to be frequent (see below).
–
Regulated Intramembrane Proteolysis (RIP) was initially identified as a new form of membrane-to-nucleus signaling mechanism. Instead of propagating signals through a cascade of intermediate messengers, transmembrane receptors directly respond to stimuli by undergoing RIP. RIP describes an evolutionary conserved mechanism that consists in the cleavage of transmembrane proteins within the plane of the membrane to liberate biologically active cytosolic fragments that enter the nucleus to control gene transcription [19][20]. Another consequence of this mechanism would be also the degradation of the protein substrate [21]. RIP proceeds in two essential steps. First, the extracytosolic (luminal or extracellular) domain is removed by the action of sheddases, principally ADAM 10 and 17 at α sites. Then, the secondary cleavage requires the inter- vention of multiprotein complexes like γ-secretase, able to hydrolyze proteins in the hydrophobic environment of the membrane bilayer [19].
–
Also, EpCAM has been identified, among many other transmembrane receptors, to undergo RIP [22]. The first cleavage, mediated by ADAM17, which is also called TACE for TNFα Converting Enzyme, triggers the release of the soluble fragment EpEX in the surrounding environment where it could act in a juxtacrine manner as a homophilic ligand for non-cleaved EpCAM [23]. Note that cell-to-cell contact is also another initial trigger for RIP of EpCAM [23]. The second cleavage by γ-secretase complexes occurs at two distinct ε- and γ-sites and respectively lead to soluble extracellular Aβ-like fragments and intracellular domain EpIC release in the cytosol of the cell. Tsaktanis et al. have recently identified by mass spectrometry the precise position of the cleavage sites of human EpCAM [24]. Although? the functions of the Aβ-like fragment are still unknown, EpIC plays a central role in downstream signalization of EpCAM (Figure 2).
–
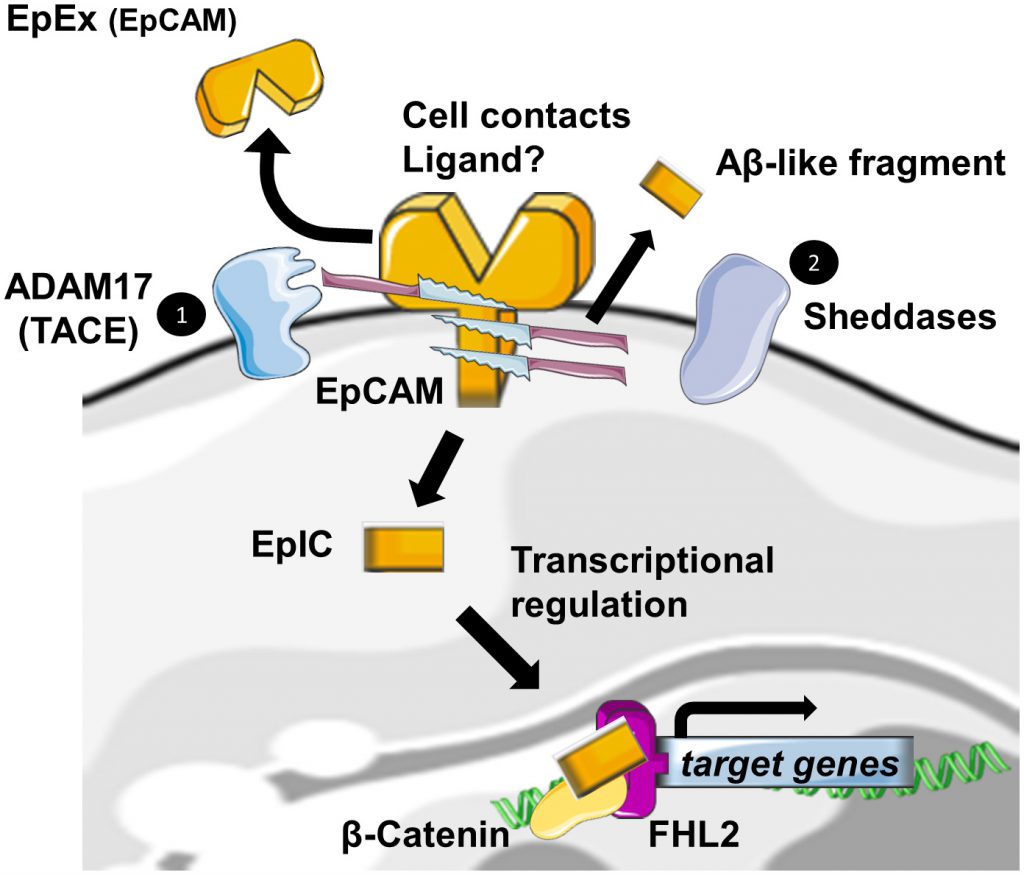 | FIGURE 2: EpCAM cleavage and downstream signalization. Cellular contacts, binding of EpEX or another unknown lig- and, lead to the activation of EpCAM cleavage by disintegrin and metalloprotease (ADAM17), and the subsequent release of the soluble EpEx domain in the intercellular space. In a second step, sheddases are acting at several sites in the transmembrane domain and generate Aβ-like fragments and an intracellular domain EpIC. If EpIC has been shown as a signalisation molecule, the functions of Aβ-like fragments are still unknown. |
–
According to the extracellular domain structure, ε- and γ -sites are always exposed in the dimeric state of the protein, whereas α sites involved in initiation of RIP and Arg80-Arg81 do not seem to be easily accessible to proteases. They are indeed directly involved in cis-dimerization interactions or sterically hindered by the glycan chains attached to Asn74 and Asn111. As only a fraction of total EpCAM is cleaved, Pavsic et al. suggested the existence of a dynamic equilibrium between monomeric and dimeric EpCAM, conformational changes induced by external yet unknown ligands of EpCAM or by sheddases themselves. The monomer–dimer equilibrium could be affected by various factors, such as lipid composition or association with other proteins [11]. BACE1 is a sheddase that has already been identified in EpCAM cleavage. Because of its optimum at pH 4.5 for enzymatic activity, BACE1 is functional in acidic intracellular compartments, including the trans-Golgi network and endosomes [25]. As full exposure of the cleavage site in the protein is achieved with the destabilization of the EpCAM dimer by a pH drop, shedding through BACE1 was suggestive of an internalization of EpCAM into acidic intracellular compartments through endocytosis. Tsaktanis et al. demonstrated this phenomenon and its dependence on clathrin proteins [24].
–
Regulation of EpCAM expression
EPCAM gene expression is controlled on the transcriptional level. The proximal promoter region of human EPCAM that predominantly controls gene transcription specifically mostly in epithelial tissues has been cloned and many transcriptions factors binding sites within this sequence have been reported so far [26]. The sequence upstream of the transcription start site (TSS) has been defined and in silico analysis of the EpCAM promoter revealed the lack of typical TATA and CAAT boxes but the presence of eukaryotic promoter elements such as initiator consensus sequences and GC boxes, as well as consensus binding sequences for transcription factors like SP-1, activator protein 1 (AP-1), activating protein 2 (AP2), Ets, ESE-1 and E-pal-like transcription factors, which are known to play a role in epithelial specific expression [26]. However, little biological data supports an actual role for these transcription factors in EPCAM gene expression. In metastatic lymph nodes from lung, breast and pancreas cancers, the upregulation of Ets family transcription factor Esx/Elf3 in metastatic lymph nodes correlated well with expression of EpCAM [4]. In ovarian cancer, Van der Gunt et al. confirmed binding of several transcription factors (AP2α, Ets1, Ets2, E2F2, E2F4 and STAT3) within the EPCAM gene by chromatin immunoprecipitation [27]. Moreover, also the tumor suppressor gene p53 was identified as a repressor of EpCAM expression and by chromatin immunoprecipitation assay, the binding of wild type p53 to a site located within intron 4 was confirmed [28]. Lastly, transcription of EPCAM was shown to be activated by TCF/β-catenin pathway via the identification of two TCF binding elements in the EPCAM promoter that specifically bound to TCF-4 [29]. Since the intracellular domain of the EpCAM protein (EpIC) can directly interact with the TCF/β-catenin protein complex, this may create a positive-feedback loop on EpCAM expression at the level of gene transcription [22], which still needs to be proven on the experimental level.
–
So far, few microRNAs controlling EPCAM mRNA expression have been identified. MicroRNA-181 has been shown to upregulate EPCAM gene expression, possibly via a positive feedback loop between miR-181 and Wnt/β-catenin signaling [30]. In prostate cancer, miRNA200c and miRNA205 were shown to induce expression of EPCAM mRNA and protein [31]. However, whether it is a direct or indirect mechanism is not known. Nevertheless, to better understand and monitor tumor cell dissemination, the identification of transcription factors or of microRNAs that govern EPCAM gene expression and that are implied in Epithelial-Mesenchymal Transition (EMT) is of high interest in the context of tumor diagnosis, as outlined below.
–
Already in 1994, it was described that EPCAM gene expression is also controlled on the epigenetic level. It was shown that DNA methylation could prevent amplification of a transfected EPCAM gene and this mechanism was suggested to occur in tumor cells [32]. Interestingly, it was confirmed almost ten years later that mutations of TP53 induce loss of DNA-methylation and amplification of the EPCAM gene [33]. However, whether DNA methylation of EPCAM gene influences DNA amplification via a replication or recombination dependent mechanism has not been identified. Together with the results that loss of p53 could also enhance EpCAM expression at the transcriptional level, these results could provide interesting mechanistic clues to understand EpCAM overexpression in cancer.
–
On the other hand, DNA methylation that occurs mainly on CpG islands of the promoters of the genes generally lead to their transcriptional silencing. Some studies have since then investigated the methylation of the promoter in various cancer types. In breast cancer, DNA hypomethylation did not correlate to tissue expression [34]. In the contrary, in ovarian, oral squamous cell carcinoma, colon and lung cancers, expression of EpCAM was correlated with DNA methylation in tissues from cancer patients. For a more complete review on DNA methylation regulation of EpCAM, see [35].
–
Two studies reported on enzymes and histone modifications involved in epigenetic regulation of the EPCAM gene. Chromatin immunoprecipitation revealed an association of repressive epigenetic marks and methylation within the EPCAM promoter increased gradually as EPCAM expression decreased in three lung adenocarcinoma cell lines [36], whereas in ovarian cancer positive cell lines, epigenetic marks that indicate activated gene transcription were immunoprecipitated with EPCAM promoter sequences [37].
–
The histone acetyl transferase p300/CBP was furthermore shown to contribute to repression of EPCAM gene expression in response to TNFα stimulation. Mechanistically, TNFα stimulation led to the activation of the transcription factor NF-kB, which then recruits p300/CBP and thereby could compete for this limited pool of cotransactivators [38].
–
EpCAM expression in healthy tissues
Tissue distribution of EpCAM has widely been investigated by immunohistochemical staining [39][40]. A strong positive signal, mainly concentrated to lateral and basal membranes, was obtained for most epithelial cell types throughout the body but not in any non-epithelial tissue like lymphoid origin and bone marrow-derived cells, mesenchymal, muscular or neuroendocrine tissue. Expression levels of EpCAM vary between different organs and cell types. In adults, epithelia of the colon, small intestine, pancreas, liver, gall bladder and endometrium owns the highest expression [39]. In general, EpCAM expression is positively correlated with proliferative and negatively correlated with more differentiated areas. One example is the epithelium of the intestine of the rat, in which a decreasing EpCAM gradient can be observed from crypts to villi, corresponding to high EpCAM expression in the intestinal stem cells which are located in the crypts and decreasing levels in the differentiated cells at the top of the villi [41]. Progenitor cells of skin epithelium express EpCAM, whereas differentiated keratinocytes do not [37]. In liver, EpCAM expression has also been observed in the precursor stem cells during regeneration processes, sustaining its role as an epithelial stem cells marker.
–
EpCAM expression in cancer
In the majority of cancer tissues, EpCAM is frequently overexpressed [39]. In contrast, the majority of squamous cell carcinomas show lower EpCAM expression than adenocarcinomas and EpCAM was found to be absent in sarcomas, lymphomas, melanomas, and neurogenic tumors [39][40]. Especially high abundant levels of EpCAM expression can be observed in carcinomas derived from colon, intestine, breast, lung and prostate. Contrary to healthy epithelia, the distribution of EpCAM varies depending on the type of carcinoma, from a basolateral to a homogenous whole cell membranous distribution. Additionally, strong EpCAM signals can also be detected in the cytoplasm and nuclei, since EpCAM is subject to regulated proteolytic cleavage [39].
–
The prognostic value of EpCAM expression is dependent on the cancer type. In some carcinoma types (thyroid, renal clear cell, head and neck squamous cell carcinomas), EpCAM immunostaining has been associated with improved survival [42][43], whereas in other carcinoma types like pancreas, bladder, gall bladder, gastric, NasoPharynx Carcinoma, EpCAM expression is associated with decreased survival [44][45]. Interestingly, for colorectal, ovarian, lung and breast carcinomas both roles have been reported [27][43]. Thus, it seems that impact of EpCAM expression is context dependent. In breast cancer for instance, EpCAM is associated with an unfavorable prognosis in the luminal and basal-like intrinsic subtypes but with a favorable prognosis in the HER2 intrinsic subtype [46].
–
To re-evaluate clinical relevance of EPCAM gene expression we utilized in a large published breast cancer cohort by using the Breast Cancer Gene-Expression Miner v4.1 [47]. In this analysis, we found a gradual rise of ECPAM gene expression related to increasing tumor grades and prognostic index status (Figure 3), indicating an increase in EPCAM expression during breast cancer progression. For more detailed reviews on the prognostic value of EpCAM expression in cancer, see [43].
–
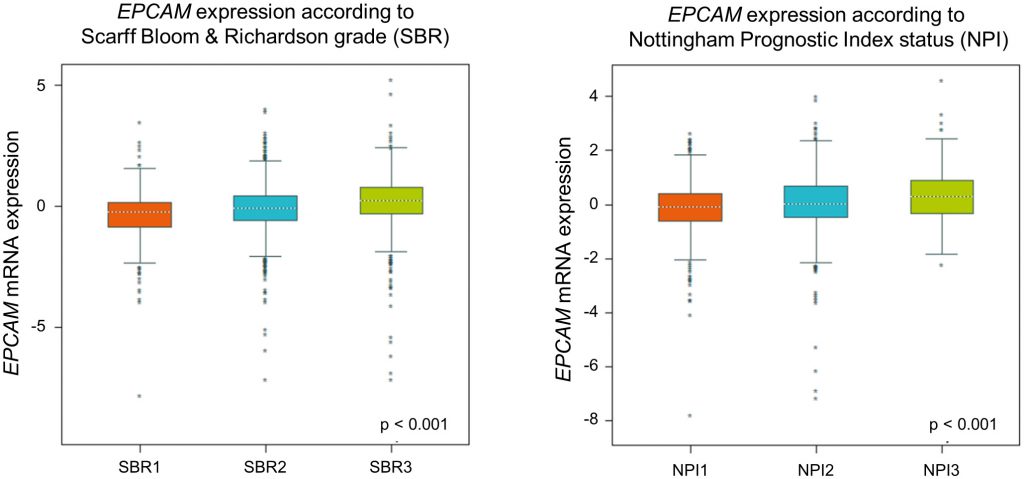 | FIGURE 3: Targeted expression analysis of normalized gene expression values of EPCAM in groups of Scarff Bloom & Richardson grade status (SBR) and Nottingham Prognostic Index status (NPI) using the Breast Cancer Gene-Expression Miner v4.1 (bc-GenExMiner v4.1). |
–
Nevertheless, an important detail is that most of these studies do not distinguish between expression and localization of the extracellular and intracellular EpCAM domains. Given the different roles of these domains, this distinction could be useful to better understand the role of EpCAM in tumorigenesis. In a retrospective study comparing oral squamous cell carcinoma, oral dysplasia and normal tissue, immunohistochemical analysis of nuclear and cytoplasmic Ep-ICD and EpEx was correlated with worse disease outcome for oral dysplasia patients [48]. In thyroid carcinomas, nuclear Ep-ICD accumulation predicted poor prognosis and was elevated in patients with anaplastic tumors [49]. In a retrospective study on breast cancer, tissues were analyzed by immunohistochemistry to determine the expression patterns of nuclear and cytoplasmic Ep-ICD and membranous EpEx and correlated with clinicopathological parameters and follow up. Nuclear Ep-ICD was identified as the most significant predictive factor for reduced disease-free survival in patients suffering from invasive ductal carcinoma. The high recurrence of disease in nuclear Ep-ICD positive patients, especially those with early tumor stage suggests that nuclear Ep-ICD accumulation holds the promise of identifying early stage patients with aggressive disease who are likely to need more rigorous post-operative surveillance and/or treatment [50]. Munz et al. have also discovered that differential glycosylation patterns of the EpCAM protein can discriminate normal from malignant tissues. EpCAM was shown to be hyperglycosylated in carcinoma tissue as compared with autologous normal epithelia. Interestingly, glycosylation of EpCAM at asparagine198 was shown to be crucial for protein stability as shown by mutagenesis of EpCAM substitution of asparagine198 for alanine led to decreased overall expression and half-life of the molecule at the plasma membrane, which is of considerable importance with respect to EpCAM variants expressed in normal and cancer tissue [51].
–
Cancer-related functions of EpCAM
Role in intercellular adhesion
Adhesive interactions of cells play an important role in the establishment and maintenance of tissue architecture. Based on their characteristic domain structure, the majority of cell surface adhesion molecules can be grouped into four families: cadherins, integrins, selectins, and cell adhesion molecules (CAMs) of the immunoglobulin (Ig) super-family. EpCAM does not belong to either of the four major families of CAMs, but was first established to mediate Ca2+-independent homophilic intercellular adhesions when introduced in cells that lack their own means of cell-cell interaction [52][53]. EpCAM was not associated with any classical junctional structures but would promote cell-cell contact via the formation of intercellular trans-oligomers [54][55]. On the molecular level EpCAM interacts with different adhesion proteins like CD44, Claudins and E-Cadherin [56].
–
Inconstant results, i.e. the cleavage or knockout (KO) of EpCAM does not affect cell adhesion [24], reported since then, recently led Lenarcic and colleagues to reinvestigate its cell adhesion property and its oligomerization capability via various analytical techniques [57]. Their data demonstrate that both EpCAM and EpEX could form cis-dimers in vitro and in vivo, but no notable higher-order oligomerization. Moreover, EpCAM molecules from adjacent cells do not form inter-cellular higher-order homo-oligomers, suggesting for the authors that EpCAM's function as a homo- philic CAM was highly implausible [57]. Nevertheless, even without direct involvement in formation of cell-cell contacts, EpCAM was initially considered to function as cell-cell adhesion protein thanks to its intracellular domain interaction with the actin cytoskeleton via α-actinin [58]. It also interacts with several important CAMs and regulates adhesive structures between cells and cell-matrix, including TJs, AJs, desmosomes, and hemi-desmosomes.
–
It was shown that EpCAM can modulate the strength of cellular adhesion mediated by E-Cadherin by disrupting the link between α-catenin and F-actin [59]. However, another study reports opposite results by showing an increase in total cellular α-catenin following EpCAM down-regulation, leading to an improved anchorage of the E-Cadherin/α-catenin/β-catenin complex to the cytoskeleton [60]. EpCAM also modifies tight junctions'composition and functions by regulating amounts and locations of claudins via a direct interaction of its transmembrane domain with claudin-7 [61][62]. These results have not been fully confirmed in viable EpCAM KO animal models yet. While a study by Lei et al. found dysfunctional tight junctions in a CTE mouse model with EpCAM inactivation [63], in a study by Guerra et al. a similar mouse model of CTE points out that adhesion junctions and E-Cadherin/beta-Catenin are affected by EpCAM loss [15]. For an exhaustive review of EpCAM's role in adhesive structures, see [64].
–
Interestingly, association of EpCAM, E Cadherin and integrin αvβ6 on tumor cells can also play additional role and trigger tumor-mediated fibroblast activation, thereby influencing both gene expression and tumor response to therapeutic agents [65].
–
Role of EpCAM in cell proliferation
The first hypothesis that EpCAM can play a role in regulating cell proliferation came from afore mentioned observation in healthy tissues that EpCAM is preferentially localized to proliferative areas [3]. Additional evidence for an active role of EpCAM in regulation of cell proliferation came from the positive correlation between EpCAM expression and cell proliferation observed in epithelial and fibroblastic cell lines [66]. Treatment of human colon and lung cancer cell lines with the specific EpCAM antibody G8.8 showed a dose-dependent increase in proliferation and revealed that most deregulated genes were involved in cell cycle regulation (like LATS2, FOSL2 and PIM1), proliferation, cell growth, apoptosis (mainly GADD45 and PIM1) and other cancer related processes [67]. EpCAM siRNA treatment resulted in a significant decrease in cell proliferation in breast cancer cell lines [60]. Tumors expressing EpCAM implanted in immunodeficient mice were furthermore strongly positive for the proliferation marker Ki67 [22]. Moreover, expression levels of EpCAM correlate with de-differentiation and malignant proliferation of epithelial cells. In 1996, Litvinov already noticed that both the level of EpCAM expression and the number of positive cells increased with the grade of carcinogenesis in cervical intraepithelial neoplasia [68]. In patients suffering from gastric cancer, high EpCAM expression has also been linked to proliferation, assessed by Ki67 staining [44].
–
Mechanistically, EpIC the soluble intracellular domain of EpCAM constitutes the signaling active intracellular compound. It is found in a large nuclear complex together with FHL2, β-catenin and Lef-1. Importantly, in this complex, FHL2 might act as a scaffold protein and links EpCAM to the Wnt pathway via interactions with β-catenin and Lef-1. This nuclear complex then binds promoters of genes involved in cell cycle regulation like c-MYC, cyclin A and E [69]. It is still unclear whether EpCAM can directly activate components of the cell cycle machinery or if EpCAM-mediated proliferation is a secondary effect of e.g. MYC upregulation, repression of apoptosis, elevation of cell metabolism or interruption of anti-proliferative signals [70]. In 2013, also Chavez-Perez et al. showed that EpCAM controls cell cycle progression via the regulation of the key player cyclin D1 at the transcriptional level [70]. There is also more recent evidence that the soluble EpEx can sustain cell proliferation by acting as a ligand of EGFR in head and neck squamous cell carcinomas and colorectal cancers by inducing cell signalization through ERK1/2 [42][71] and AKT [71].
–
Role of EpCAM in stemness
EpCAM is mostly expressed in epithelial cells but likewise also expressed in various tissue stem cells, precursors, and in murine and human embryonic stem cells [69][72], which has important implications for cancer progression. EpCAM expression is tightly regulated at earliest time points of gastrulation in order to achieve a mandatory spatiotemporal cellular heterogeneity of EpCAM in endo-and mesodermal lineages [73]. It has also been associated with morphogenesis based on the marked variations of its expression during development and regeneration of epithelia. In later stages of epithelial development, EpCAM acquires a strictly epithelial-specific expression, whereas in terminally differentiated cells EpCAM is not expressed [3][74][75]. For example, in liver, EpCAM was only detected in regenerating cells like hepatobiliary stem and progenitor cells, while it was lost in mature hepatocytes.
–
Key signals implicated in stem cell phenotype are provided by components of the Wnt pathway, LIF/STAT3 and c-Myc, and by the transcription factors Nanog, Oct3/4, Klf4, and Sox2. Indeed, these factors play a central role to the conversion of somatic cells into induced pluripotent stem cells [76]. Mechanistically, EpIC and the Wnt pathway are linked to each other, notably via FHL2 scaffold protein (see above). Moreover, they have also been demonstrated to be direct targets of EpCAM in human embryonic stem cells [77]. Recently, Kuan et al. suggested that EpEX and EpCAM could also trigger reprogramming of fibroblasts into induced pluripotent stem cells via activation of STAT3 [78].
–
Cancer stem cells share many similar biological properties with embryonic stem cells. Precisely, cancer stem cells, like embryonic stem cells, undergo molecular regulations such as persistent self-renewal. EpCAM has been used in combination with CD44 as a marker to efficiently isolate cancer stem cells in different cancer entities such as colon, breast, pancreas and prostate carcinomas [79]. Especially in tumor-initiating cells from the liver, an important function for the EpCAM protein has been well described [29]: an interrelation of EpCAM and Wnt in hepatocellular carcinomas was sustained with the finding that the EPCAM gene becomes transcriptionally activated by TCF-4, a member of the Lef family of transcription factors [29]. Taken together, these findings depict an important role for EpCAM in the induction and/or maintenance of proliferation and cellular differentiation of progenitors, stem cells, induced pluripotent stem cells, cancer cells, and cancer stem cells. Stem cell phenotypes and mesenchymal characters have often been conflated [80][81]. Interestingly, some studies also report a parallel regulation of reprogramming factors and EMT-TF by EpCAM [45][82].
–
EpCAM and Epithelial to Mesenchymal Transition (EMT)
EMT is nowadays described as a complex program, governed by specific transcription factors, miRNA, epigenetic and post-translational regulators and executed in response to pleiotropic factors that leads to the modification of the adhesion molecules expressed by the cell and the further acquisition of a migratory and invasive behavior [83]. Contrary to E Cadherin, biological regulation of EpCAM during EMT seems to be less well established. Therefore, in the following paragraph, we will present the different studies that have addressed this pivotal question.
–
In 1998 Jojovic et al. were the first to suggest that EMT leads to a transient loss of EpCAM expression during the migratory and early pro-migratory period by evaluating the expression at EpCAM in immunohistochemistry on breast and lung cell lines implanted in immunocompromised mice [84]. Then, gene expression analyses demonstrate that EpCAM is decreased in mesenchymal-like cancers. In breast cancer, EpCAM was down-regulated in mesenchymal lines relative to the epithelial cell lines [85] and in EMT-induced breast cancer cells [86][87]. EpCAM was also one of the downregulated genes in an EMT gene signature developed from NSCLC cell lines [88]. In addition, a negative correlation between the activity of EMT-associated transcription factors SNAI1 [89], Slug [90] and ZEB1 [31] and EpCAM expression has been reported. Nevertheless, a direct functional link between EMT and EpCAM expression was missing. To directly investigate the impact of EMT on EpCAM expression, normal epithelial and various epithelial cancer cell lines were treated in vitro with transforming growth factor-β1 (TGFβ1) and tumor necrosis factor-α (TNFα), a combination that is known to induce EMT [91]. Following 72 h of cytokine treatment, immunofluorescence staining of cells showed decreased expression of the epithelial markers EpCAM and E-cadherin and increased expression of the mesenchymal marker vimentin compared with control cells. Moreover, growth factor stimulation leading to ERK2 activation (a key regulator of EMT) suppressed EpCAM expression. Similar downregulation of EpCAM at mRNA and protein levels was obtained in lung and esophageal cell lines after treatment with TGF [92]. More specifically, ERK2 suppresses EpCAM transcription through activation of EMT-associated transcription factors SNAI1, SNAI2, TWIST1 and ZEB1, which bind to E-box sites in the EpCAM promoter [91]. These results are in line with previous evidence that ZEB1 directly binds to the EPCAM promoter, leading to a ZEB1-dependent repression of EPCAM expression in human pancreatic and breast cancer cell lines [93].
–
Taken together, these results are in favor of a downregulation of EpCAM expression during EMT. Interestingly, EpCAM could also contribute to the regulation of EMT by suppressing ERK activity and SNAIL2 expression, defining a double-negative feedback loop (see above) [91]. As several double-negative feedback loops have been described in the regulation of EMT, notably feedback loops involving miR let-7 and LIN28, miR15a/16-1 and AP4, miR-34 and SNAI1 and miR-200 and ZEB1 [83], this double-negative feedback loop for EpCAM may be particularly noteworthy in the regulation of EMT in epithelial cancers. Furthermore, soluble EpEX was demonstrated as a ligand that induces specific activation of classical EGFR signaling pathways. Since EGFR signaling pathways promote EMT-characteristic phenotypic changes, the authors addressed whether EpEX could modulate EGF-dependent EMT in HNSCC cell lines and showed that soluble EpEX-Fc, acting as an EGF competitor for EGFR, counteracted EMT via repression of the EMTTF (Snail, ZEB1, and Slug) activation. This mechanism provides additional insights on the juxtacrine role of EpEx domain, shed in the tumor microenvironment after RIP of EpCAM (Figure 4) [42].
–
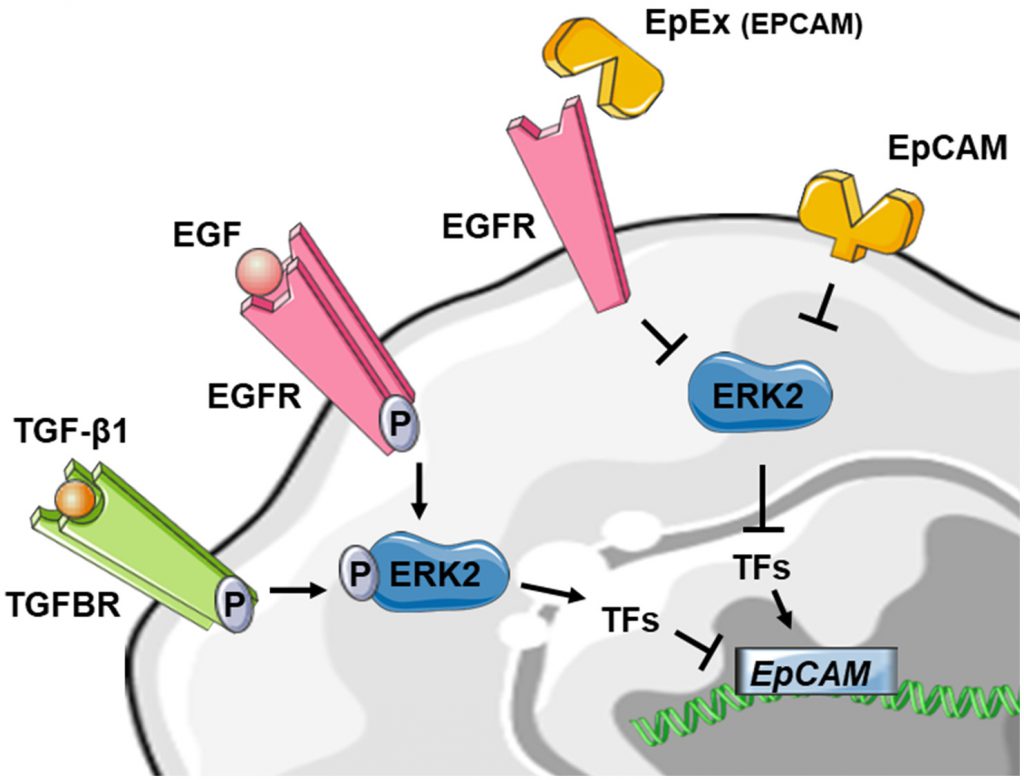 | FIGURE 4: Reduced schematic of EpCAM regulation in the context of EMT. Stimulation of tyrosine kinase receptors via TGF-β1 or EGF is leading to ERK2 activation and suppresses EPCAM mRNA expression indirectly through activation of EMT-associated transcription factors (TF) like SNAI1, SNAI2, TWIST1 and ZEB1. The EpCAM protein on the other hand could also contribute to the regulation of EMT by suppressing ERK2 activity. The soluble EpEX-fragment is acting as an EGF competitor for EGFR, counteracted EMT via inhibiting of the EMTTF (Snail, ZEB1, and Slug) which leads to activation of EPCAM expression. For detailed review, see references [42][91][92]. |
–
On the other hand, some studies have suggested a promoting role in EMT for EpCAM. Indeed, it has first been noticed that knockdown of EpCAM could inhibit the expression of EMT-transcription factors Snail and Slug in colon cancer [82] and that its overexpression could enhance TGF-β1-induced EMT in MCF-7 breast cancer cells [94]. More recently, in nasopharynx carcinoma, the mesenchymal markers N-cadherin, Vimentin, β-catenin and the EMTTF Slug were significantly upregulated, whereas the epithelial markers α-catenin and E-cadherin were decreased in the EpCAM expressing cells. Notably, this regulation involved the PTEN/AKT/mTOR pathway [45].
–
Therefore, regulation of EpCAM during EMT seems to be context dependent. As it is well established that cells during EMT no longer oscillate between full epithelial and full mesenchymal states but, rather, present high plasticity and can adopt a spectrum of intermediary phases [83]. Lineage tracing experiments in animal models to measure EpCAM expression in disseminating cells bearing different states in EMT transition would greatly help to confirm or infirm these results obtained in two dimentional cell culture experiments [83].
–
EpCAM role in invasion/migration
It can be, in a first sight, counter intuitive that a supposed adhesion molecule like EpCAM can promote the mobility of cells and tissues. However, implication of EpCAM in regulation of cell migration was studied during Xenopus gastrulation, a model to study morphogenetic movements [95]. EpCAM levels crucially regulate movements of cells in embryonic tissues via its EpIC domain acting as an inhibitor of a novel PKC isoform. Furthermore, in a conditional KO mouse model, loss of EpCAM impairs the migration of skin Langerhans cells [96]. On the opposite, migration of enterocytes in defective EpCAM mutant mice present significantly higher migration rates compared to wild type mice [14]. These data are noteworthy as acquisition of a migratory/invasive phenotype is also intrinsically linked with EMT. However, results on cancer cell lines about EpCAM are also conflicting and seem to be context dependent. Strong EpCAM overexpression was associated with enhanced invasion of breast cancer cell lines into extracellular matrix [60] and consisting results were observed upon silenced EpCAM expression due to the binding of the tumor suppressor p53 to promoter elements of the EPCAM gene [28]. In a subsequent study, the same team showed that the transcription factor activator protein 1 (AP-1) is an important downstream mediator of EpCAM signaling in breast cancer biology through the MEKK1-MKK7-JNK cascade [97]. Another way for EpCAM to promote invasion of breast cancer cells (ER negative) would be via upregulation of IL8 expression (a member of the CXC chemokine family associated with increased breast cancer invasion in vitro) but a precise molecular mechanism has not been identified yet [98]. However, specific ablation of EpCAM was also reported to increase invasion and migration in MCF-10A cells, underlying the importance of context dependence in our understanding of EpCAM signalization [91]. Direct involvement of EpCAM in the migratory phenotype of esophageal carcinoma was tested upon siRNA-mediated downregulation of EpCAM. An increased migration capacity of cells after EpCAM knock-down was paralleled by an increase of vimentin expression [92]. Moreover, in migrating esophageal carcinoma cells and in head and neck carcinoma cells, a progressive loss of EpCAM expression occurs at the membrane with the appearance of EpCAM positive speckles in the cytoplasm, suggesting EpCAM endocytosis and degradation [24][92].
RELEVANCE OF EpCAM AS A DIAGNOSTIC MARKER IN LIQUID BIOPSY ASSAYS
EpCAM expression in CTC
Targeting EpCAM to capture CTC was a proven successful strategy by numerous clinical CTC studies on thousands of cancer patients demonstrating the prognostic relevance of EpCAM (+) CytoKeratins (+) CTCs in breast, prostate, lung or colorectal cancers, and other epithelial tumors, as reviewed elsewhere [99][100]. Recently, the prognostic value of these cells in early stage breast cancer patients receiving neoadjuvant therapy was confirmed in a large meta-analysis [101]. Most of these clinical studies are based on the use of the CellSearch system that proceed with a ferromagnetic EpCAM-based capture of cells followed by their phenotypic characterization (cytokeratin (+), DAPI (+) CD45 (-)). This CTC assay has been extensively validated for its analytical accuracy and reproducibility [102]. Different EpCAM antibodies clones result in different CTC capture yield [103]. Thus, the question whether different antibodies binding different epitopes on the protein could improve recovery yield need further investigation [104].
–
However, the presence of EpCAM (-) CTCs in the blood circulation [105] has been also reported for patients with breast cancers [106][107], colorectal cancer [108], or non small cell lung cancer [109]. Moreover, it was experimentally investigated in a mouse model that CTCs could escape from EpCAM-based detection due to EMT [110]. Together with the supposed necessity of EMT as a prerequisite for cancer dissemination, these results have raised doubt whether EpCAM-based capture is sufficient to escape all CTCs relevant to metastatic progression.
–
Therefore, many studies have focused their attention on CTCs with pronounced mesenchymal traits, expressing N-Cadherin, O-Cadherin, vimentin and fibronectin and some could find a correlation with clinical parameters like higher disease stage, presence of metastases, therapy response and worse outcome [111]. Nevertheless, there is no broadly used and established marker for specific enrichment of CTC with mesenchymal phenotype to date. Several approaches have been introduced, but none of them has been conclusively proven to enable a specific enrichment of cancer cells. Mesenchymal markers such as N-Cadherin and vimentin are frequently expressed on PBMCs (peripheral blood mononuclear cells). Therefore, these markers are not suitable for antigen-dependent enrichment of CTC and antigen-dependent enrichment techniques allowing a specific enrichment of CTC with mesenchymal phenotype have not been broadly implemented yet [112]. An alternative approach is to perform a negative selection for leukocyte markers in combination with the detection of mesenchymal markers [112].
–
To specifically capture CTCs with low or missing expression of EpCAM, additional epithelial specific cell surface markers, like EGFR, HER2, MUC1 [113][114], have been applied to increase the sensitivity of CTC detection. Another option is the search for novel markers that are not downregulated by CTCs during their EMT and not expressed in blood cells. On the basis of comparative micro-array analyses, the PLS3 gene was identified, which codes for the Plastin-3 protein, an actin-bundling protein known to inhibit cofilin-mediated depolymerization of actin fibers. Plastin-3 was demonstrated as a suitable new marker for CTCs in patients with colorectal cancer, especially in early-stage patients [115]. More recently, the recombinant VAR2CSA protein was found to bind a uniquely modified form of chondroitin sulfate, which is expressed by placental cells and cancer cells of both epithelial and mesenchymal origin. Its implementation in dedicated capture device led to a markedly enhanced CTC capture compared to EpCAM based method with the capacity to capture additional EpCAM negative CTC [116].
–
Another consequence of the drawbacks of EpCAM-based enrichment was the development of new label-free technologies for enrichment and detection of CTCs. These novel approaches are mainly based on the assumption that CTCs have different physical properties (size, deformability, density) than the surrounding blood cells. However, it can be assumed that these physical parameters might be also affected by EMT and EpCAM expression [87][117].
–
Other studies have investigated the clinical relevance of cells that have not been captured by Cellsearch. These cells were captured from Cellsearch discard by filtration and identified with immunofluorescence staining against cytokeratin expression [118][119]. Significant additional CTC numbers were identified with filtration in lung, breast and cancer patient samples. Clinical correlation with a worse outcome was found with EpCAM + CTC whereas EpCAM – CTC had no relation with overall survival. If the EpCAM – cells that have been collected are proven as tumorous, these results highlight the need to better discriminate the different populations of CTC regarding EpCAM expression.
–
Notably, concomitant detection of epithelial and mesenchymal markers has been reported within EpCAM+ CTC [120][121]. Assessment of keratin expression as epithelial marker has also been successfully used in this context [122]. These results corroborate previous findings by Yu et al. demonstrating that a significant proportion of CTC bears a hybrid epithelial/mesenchymal phenotype [123] and are in line with the actual conception of EMT as a continuum, and not a binary switch between two extreme phenotypic stages [83]. It is also noteworthy here that the cells bearing the most mesenchymal phenotypes would not be the ones that present the highest metastatic capacity [83].
–
Taken together, despite the obvious shortcoming that EpCAM-based CTC detection might miss CTCs that have undergone EMT, EpCAM still outperforms other tested surface markers for CTC enrichment and detection, which is indicated by the fact that the CellSearch system still remains the only FDA-cleared technology for CTC detection. Better knowledge of the range of expression of EpCAM in CTC from patients and comparison of the clinical relevance of the different CTC populations with differential EpCAM expression could help to solve this ongoing debate.
–
EpCAM in Extracellular Vesicles
Extracellular Vesicles (EVs) have emerged during the last ten years as critical mediators of cell-cell communication, involved in many normal physiological processes as well as in cancer progression. EVs can be isolated from bodily fluids including blood, urine, breast milk, ascites, bronchoalveolar lavage fluids and transport diverse nucleic acids (DNA, RNA, microRNA) and proteins as part of their function in intercellular communication [124]. Therefore, EVs might be interesting candidates as biomarkers for monitoring tumor evolution or response to therapy.
–
EVs is a general term to virtually describe any type of membrane particle released by any type of cell, into the extracellular space, regardless of differences in biogenesis and composition. Current criteria to distinguish between diverse EV populations are based on size, density, subcellular origin, function and molecular cargo. Based on a size, different categories can be distinguished: exosomes (30-100 nm diameter), microvesicles (MVs) (100-1000 nm diameter), and a more recently identified cancer-derived EV population termed “large oncosomes,” which are much larger than most EV types characterized to date (1-10 μm diameter) [125][126].
–
EVs are highly enriched for tetraspanins, a protein superfamily that organize membrane microdomains termed tetraspanin-enriched microdomains (TEMs) by forming clusters and interacting with cholesterol and gangliosides and a large variety of TM and cytosolic signaling proteins [127]. Importantly, EpCAM has been associated to TEM via interaction with different tetraspanin proteins (CD9, CO-O29 as well as CD44 variant isoform) [128]. It has been recently demonstrated that EpCAM is essential for the gastrointestinal localization of some EVs secreted from the intestinal epithelia cells and implicated in the intestinal tract immune balance [129]. Interestingly, a proteomic analysis of the EV content from cancer cell lines, led Tauro et al. to define a distinct population of exosomes according to EpCAM expression [130] and showed colocalization of EpCAM with CD44 and claudin 7, proteins that are known to complex together to promote tumor progression [130]. In ovarian cancer patients, Im et al. detected the expression of EpCAM on exosomes from ascites at higher levels than in the control group of noncancerous ascites from cirrhosis patients [131]. More recently, EpCAM was also detected in exosomes fraction after surface protein exosomal profiling in plasma from pancreatic cancer patients [132]. Further investigations are necessary to confirm these data suggesting that EpCAM in circulation may represent a cancer-specific or at least cancer-associated exosomal biomarker.
–
Detection of soluble forms of EpCAM in blood serum of cancer patients has reported disappointing results, notably due to a lack of correlation of with important clinical end-points like overall survival in most studies [133][134][135][136][137][138][139]. Nevertheless, it might be interesting to compare the biological and clinical relevance of the exosomal forms of EpCAM with soluble EpEX domain.
CONCLUSION
EpCAM expression is abundant in normal and malignant epithelial cells and the high immunogenicity of the EpCAM protein has enabled the development of specific antibodies that can be applied for immunohistochemistry analysis and enrichment of CTCs. EpCAM-based approaches for CTC detection have significantly contributed to validate the clinical relevance of CTCs in various cancer entities. Despite its widespread use as “epithelial marker”, the biological functions of EpCAM deserve more intense investigation. Besides affecting intercellular adhesion, EpCAM influences important other functions relevant to tumor progression including cell proliferation and cancer stemness, which suggests an active role of EpCAM in cancer metastasis.
–
Investigating the dynamic range of EpCAM expression on CTC in different cancer entities might provide new insights in the biology of cancer metastasis. A precise quantification of EpCAM expression in CTCs might help to better understand the prognostic role of the EpCAM+ CTCs demonstrated in numerous clinical studies. EpCAM and its signaling internal domain EpIC have been implicated in cancer stemness and the EMT process, suggesting that EpCAM is more than just a marker for CTCs. Understanding the regulation of EpCAM expression is important and in this context the methylation status of EpCAM in CTCs might provide important information. Besides CTCs, EVs isolated from the blood (and other body fluids) of cancer patients might provide complementary information. Thus, liquid biopsy analyses open new avenues to study the relevance of EpCAM for cancer metastasis and may lead to improvements in the personalized management of cancer patients.
REFERENCES
- Herlyn M, Steplewski Z, Herlyn D, Koprowski H (1979). Colorectal carcinoma-specific antigen: detection by means of monoclonal antibodies. Proc Natl Acad Sci U S A 76(3): 1438-1442. doi: 10.1073/pnas.76.3.1438
- Schnell U, Cirulli V, Giepmans BN (2013). EpCAM: structure and function in health and disease. Biochim Biophys Acta 1828(8): 1989- 2001. doi: 10.1016/j.bbamem.2013.04.018
- Trzpis M, McLaughlin PM, de Leij LM, Harmsen MC (2007). Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am J Pathol 171(2): 386-395. doi: 10.2353/ajpath.2007.070152
- Baeuerle PA, Gires O (2007). EpCAM (CD326) finding its role in cancer. Br J Cancer 96(3): 417-423. doi: 10.1038/sj.bjc.6603494
- Bardelli A, Pantel K (2017). Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell 31(2): 172-179. doi: 10.1016/j.ccell.2017.01.002
- Pantel K, Alix-Panabieres C (2010). Circulating tumor cells in cancer patients: challenges and perspectives. Trends Mol Med 16(9): 398- 406. doi: 10.1016/j.molmed.2010.07.001
- Mazel M, Jacot W, Pantel K, Bartkowiak K, Topart D, Cayrefourcq L, Rossille D, Maudelonde T, Fest T, Alix-Panabieres C (2015). Frequent expression of PD-L1 on circulating breast cancer cells. Mol Oncol 9(9): 1773-1782. doi: 10.1016/j.molonc.2015.05.009
- Carter L, Rothwell DG, Mesquita B, Smowton C, Leong HS, Fernandez-Gutierrez F, Li Y, Burt DJ, Antonello J, Morrow CJ, Hodgkinson CL, Morris K, Priest L, Carter M, Miller C, Hughes A, Blackhall F, Dive C, Brady G (2017). Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med 23(1): 114-119. doi: 10.1038/nm.4239
- Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Zhu Y, Silberstein JL, Taylor MN, Maughan BL, Denmeade SR, Pienta KJ, Paller CJ, Carducci MA, Eisenberger MA, Luo J (2017). Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J Clin Oncol 35(19): 2149-2156. doi: 10.1200/jco.2016.70.1961
- Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, Klein C, Saini M, Bauerle T, Wallwiener M, Holland-Letz T, Hofner T, Sprick M, Scharpff M, Marme F, Sinn HP, Pantel K, Weichert W, Trumpp A (2013). Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol 31(6): 539-544. doi: 10.1038/nbt.2576
- Pavsic M, Guncar G, Djinovic-Carugo K, Lenarcic B (2014). Crystal structure and its bearing towards an understanding of key biological functions of EpCAM. Nat Commun 5(4764. doi: 10.1038/ncomms5764
- Pathak SJ, Mueller JL, Okamoto K, Das B, Hertecant J, Greenhalgh L, Cole T, Pinsk V, Yerushalmi B, Gurkan OE, Yourshaw M, Hernandez E, Oesterreicher S, Naik S, Sanderson IR, Axelsson I, Agardh D, Boland CR, Martin MG, Putnam CD, Sivagnanam M (2019). EPCAM mutation update: Variants associated with congenital tufting enteropathy and Lynch syndrome. Hum Mutat 40(2): 142-161. doi: 10.1002/humu.23688
- Kozan PA, McGeough MD, Pena CA, Mueller JL, Barrett KE, Marchelletta RR, Sivagnanam M (2015). Mutation of EpCAM leads to intestinal barrier and ion transport dysfunction. J Mol Med 93(5): 535- 545. doi: 10.1007/s00109-014-1239-x
- Mueller JL, McGeough MD, Pena CA, Sivagnanam M (2014). Functional consequences of EpCam mutation in mice and men. Am J Physiol Gastrointest Liver Physiol 306(4): G278-288. doi: 10.1152/ajpgi.00286.2013
- Guerra E, Lattanzio R, La Sorda R, Dini F, Tiboni GM, Piantelli M, Alberti S (2012). mTrop1/Epcam knockout mice develop congenital tufting enteropathy through dysregulation of intestinal E- cadherin/beta-catenin. PloS One 7(11): e49302. doi: 10.1371/journal.pone.0049302
- Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, Lee TY, Bodmer D, Hoenselaar E, Hendriks-Cornelissen SJ, Tsui WY, Kong CK, Brunner HG, van Kessel AG, Yuen ST, van Krieken JH, Leung SY, Hoogerbrugge N (2009). Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet 41(1): 112-117. doi: 10.1038/ng.283
- Balzar M, Bakker HA, Briaire-de-Bruijn IH, Fleuren GJ, Warnaar SO, Litvinov SV (1998). Cytoplasmic tail regulates the intercellular adhesion function of the epithelial cell adhesion molecule. Mol Cell Biol 18(8): 4833-4843. doi: 10.1128/mcb.18.8.4833
- Maghzal N, Kayali HA, Rohani N, Kajava AV, Fagotto F (2013). EpCAM controls actomyosin contractility and cell adhesion by direct inhibition of PKC. Dev Cell 27(3): 263-277. doi: 10.1016/j.devcel.2013.10.003
- Brown MS, Ye J, Rawson RB, Goldstein JL (2000). Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100(4): 391-398. doi: 10.1016/S0092- 8674(00)80675-3
- Wolfe MS, Kopan R (2004). Intramembrane proteolysis: theme and variations. Science 305(5687): 1119-1123. doi: 10.1126/science.1096187
- Kopan R, Ilagan MX (2004). Gamma-secretase: proteasome of the membrane? Nat Rev Mol Cell Biol 5(6): 499-504. doi: 10.1038/nrm1406
- Maetzel D, Denzel S, Mack B, Canis M, Went P, Benk M, Kieu C, Papior P, Baeuerle PA, Munz M, Gires O (2009). Nuclear signalling by tumor-associated antigen EpCAM. Nat Cell Biol 11(2): 162-171. doi: 10.1038/ncb1824
- Denzel S, Maetzel D, Mack B, Eggert C, Barr G, Gires O (2009). Initial activation of EpCAM cleavage via cell-to-cell contact. BMC Cancer 9: 402. doi: 10.1186/1471-2407-9-402
- Tsaktanis T, Kremling H, Pavsic M, von Stackelberg R, Mack B, Fukumori A, Steiner H, Vielmuth F, Spindler V, Huang Z, Jakubowski J, Stoecklein NH, Luxenburger E, Lauber K, Lenarcic B, Gires O (2015). Cleavage and cell adhesion properties of human epithelial cell adhesion molecule (HEPCAM). J Biol Chem 290(40): 24574-24591. doi: 10.1074/jbc.M115.662700
- Cole SL, Vassar R (2007). The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol Neurodegener 2: 22. doi: 10.1186/1750-1326-2- 22
- McLaughlin PM, Trzpis M, Kroesen BJ, Helfrich W, Terpstra P, Dokter WH, Ruiters MH, de Leij LF, Harmsen MC (2004). Use of the EGP-2/Ep-CAM promoter for targeted expression of heterologous genes in carcinoma derived cell lines. Cancer Gene Ther 11(9): 603- 612. doi: 10.1038/sj.cgt.7700725
- van der Gun BT, de Groote ML, Kazemier HG, Arendzen AJ, Terpstra P, Ruiters MH, McLaughlin PM, Rots MG (2011). Transcription factors and molecular epigenetic marks underlying EpCAM overexpression in ovarian cancer. Br J Cancer 105(2): 312-319. doi: 10.1038/bjc.2011.231
- Sankpal NV, Willman MW, Fleming TP, Mayfield JD, Gillanders WE (2009). Transcriptional repression of epithelial cell adhesion molecule contributes to p53 control of breast cancer invasion. Cancer Res 69(3): 753-757. doi: 10.1158/0008-5472.can-08-2708
- Yamashita T, Budhu A, Forgues M, Wang XW (2007). Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res 67(22): 10831-10839. doi: 10.1158/0008-5472.can-07-0908
- Ji J, Yamashita T, Wang XW (2011). Wnt/beta-catenin signaling activates microRNA-181 expression in hepatocellular carcinoma. Cell Biosci 1(1): 4. doi: 10.1186/2045-3701-1-4
- Massoner P, Thomm T, Mack B, Untergasser G, Martowicz A, Bobowski K, Klocker H, Gires O, Puhr M (2014). EpCAM is overexpressed in local and metastatic prostate cancer, suppressed by chemotherapy and modulated by MET-associated miRNA-200c/205. Br J Cancer 111(5): 955-964. doi: 10.1038/bjc.2014.366
- Alberti S, Nutini M, Herzenberg LA (1994). DNA methylation prevents the amplification of TROP1, a tumor-associated cell surface antigen gene. Proc Natl Acad Sci U S A 91(13): 5833-5837. doi: 10.1073/pnas.91.13.5833
- Nasr AF, Nutini M, Palombo B, Guerra E, Alberti S (2003). Mutations of TP53 induce loss of DNA methylation and amplification of the TROP1 gene. Oncogene 22(11): 1668-1677. doi: 10.1038/sj.onc.1206248
- Spizzo G, Gastl G, Obrist P, Fong D, Haun M, Grunewald K, Parson W, Eichmann C, Millinger S, Fiegl H, Margreiter R, Amberger A (2007). Methylation status of the Ep-CAM promoter region in human breast cancer cell lines and breast cancer tissue. Cancer Lett 246(1-2): 253- 261. doi: 10.1016/j.canlet.2006.03.002
- van der Gun BT, Melchers LJ, Ruiters MH, de Leij LF, McLaughlin PM, Rots MG (2010). EpCAM in carcinogenesis: the good, the bad or the ugly. Carcinogenesis 31(11): 1913-1921. doi: 10.1093/carcin/bgq187
- Tai KY, Shiah SG, Shieh YS, Kao YR, Chi CY, Huang E, Lee HS, Chang LC, Yang PC, Wu CW (2007). DNA methylation and histone modification regulate silencing of epithelial cell adhesion molecule for tumor invasion and progression. Oncogene 26(27): 3989-3997. doi: 10.1038/sj.onc.1210176
- Klein CE, Cordon-Cardo C, Soehnchen R, Cote RJ, Oettgen HF, Eisinger M, Old LJ (1987). Changes in cell surface glycoprotein expression during differentiation of human keratinocytes. J Invest Dermatol 89(5): 500-506. doi: 10.1111/1523-1747.ep12460996
- Gires O, Kieu C, Fix P, Schmitt B, Munz M, Wollenberg B, Zeidler R (2001). Tumor necrosis factor alpha negatively regulates the expression of the carcinoma-associated antigen epithelial cell adhesion molecule. Cancer 92(3): 620-628. doi: 10.1002/1097- 0142(20010801)92:3<620::aid-cncr1362>3.0.co;2-f
- Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G, Dirnhofer S (2004). Frequent EpCam protein expression in human carcinomas. Hum Pathol 35(1): 122-128. doi: 10.1016/j.humpath.2003.08.026
- Momburg F, Moldenhauer G, Hammerling GJ, Moller P (1987). Immunohistochemical study of the expression of a Mr 34,000 human epithelium-specific surface glycoprotein in normal and malignant tissues. Cancer Res 47(11): 2883-2891. PMID: 3552208
- Schiechl H, Dohr G (1987). Immunohistochemical studies of the distribution of a basolateral-membrane protein in intestinal epithelial cells (GZ1-Ag) in rats using monoclonal antibodies. Histochemistry 87(5): 491-498. doi: 10.1007/bf00496823
- Pan M, Schinke H, Luxenburger E, Kranz G, Shakhtour J, Libl D, Huang Y, Gaber A, Pavsic M, Lenarcic B, Kitz J, Jakob M, Schwenk- Zieger S, Canis M, Hess J, Unger K, Baumeister P, Gires O (2018). EpCAM ectodomain EpEX is a ligand of EGFR that counteracts EGF- mediated epithelial-mesenchymal transition through modulation of phospho-ERK1/2 in head and neck cancers. PLoS Biol 16(9): e2006624. doi: 10.1371/journal.pbio.2006624
- Herreros-Pomares A, Aguilar-Gallardo C, Calabuig-Farinas S, Sirera R, Jantus-Lewintre E, Camps C (2018). EpCAM duality becomes this molecule in a new Dr. Jekyll and Mr. Hyde tale. Crit Rev Oncol Hematol 126: 52-63. doi: 10.1016/j.critrevonc.2018.03.006
- Kroepil F, Dulian A, Vallbohmer D, Geddert H, Krieg A, Vay C, Topp SA, am Esch JS, Baldus SE, Gires O, Knoefel WT, Stoecklein NH (2013). High EpCAM expression is linked to proliferation and lauren classification in gastric cancer. BMC Res Notes 6: 253. doi: 10.1186/1756-0500-6-253
- Wang MH, Sun R, Zhou XM, Zhang MY, Lu JB, Yang Y, Zeng LS, Yang XZ, Shi L, Xiao RW, Wang HY, Mai SJ (2018). Epithelial cell adhesion molecule overexpression regulates epithelial-mesenchymal transition, stemness and metastasis of nasopharyngeal carcinoma cells via the PTEN/AKT/mTOR pathway. Cell Death Dis 9(1): 2. doi: 10.1038/s41419-017-0013-8
- Soysal SD, Muenst S, Barbie T, Fleming T, Gao F, Spizzo G, Oertli D, Viehl CT, Obermann EC, Gillanders WE (2013). EpCAM expression varies significantly and is differentially associated with prognosis in the luminal B HER2(+), basal-like, and HER2 intrinsic subtypes of breast cancer. Br J Cancer 108(7): 1480-1487. doi: 10.1038/bjc.2013.80
- Jezequel P, Campone M, Gouraud W, Guerin-Charbonnel C, Leux C, Ricolleau G, Campion L (2012). bc-GenExMiner: an easy-to-use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res Treat 131(3): 765-775. doi: 10.1007/s10549-011-1457-7
- Somasundaram RT, Kaur J, Leong I, MacMillan C, Witterick IJ, Walfish PG, Ralhan R (2016). Subcellular differential expression of Ep- ICD in oral dysplasia and cancer is associated with disease progression and prognosis. BMC Cancer 16: 486. doi: 10.1186/s12885-016-2507-7
- He HC, Kashat L, Kak I, Kunavisarut T, Gundelach R, Kim D, So AK, MacMillan C, Freeman JL, Ralhan R, Walfish PG (2012). An Ep-ICD based index is a marker of aggressiveness and poor prognosis in thyroid carcinoma. PloS One 7(9): e42893. doi: 10.1371/journal.pone.0042893
- Srivastava G, Assi J, Kashat L, Matta A, Chang M, Walfish PG, Ralhan R (2014). Nuclear Ep-ICD accumulation predicts aggressive clinical course in early stage breast cancer patients. BMC Cancer 14: 726. doi: 10.1186/1471-2407-14-726
- Munz M, Fellinger K, Hofmann T, Schmitt B, Gires O (2008). Glycosylation is crucial for stability of tumor and cancer stem cell antigen EpCAM. Front Biosci 13: 5195-5201. doi: 10.2741/3075
- Litvinov SV, Velders MP, Bakker HA, Fleuren GJ, Warnaar SO (1994). Ep-CAM: a human epithelial antigen is a homophilic cell-cell adhesion molecule. J Cell Biol 125(2): 437-446. doi: 10.1083/jcb.125.2.437
- Litvinov SV, Bakker HA, Gourevitch MM, Velders MP, Warnaar SO (1994). Evidence for a role of the epithelial glycoprotein 40 (Ep-CAM) in epithelial cell-cell adhesion. Cell Adhes Commun 2(5): 417-428. doi: 10.3109/15419069409004452
- Balzar M, Prins FA, Bakker HA, Fleuren GJ, Warnaar SO, Litvinov SV (1999). The structural analysis of adhesions mediated by Ep-CAM. Exp Cell Res 246(1): 108-121. doi: 10.1006/excr.1998.4263
- Trebak M, Begg GE, Chong JM, Kanazireva EV, Herlyn D, Speicher DW (2001). Oligomeric state of the colon carcinoma-associated glycoprotein GA733-2 (Ep-CAM/EGP40) and its role in GA733- mediated homotypic cell-cell adhesion. J Biol Chem 276(3): 2299- 2309. doi: 10.1074/jbc.M004770200
- Kuhn S, Koch M, Nubel T, Ladwein M, Antolovic D, Klingbeil P, Hildebrand D, Moldenhauer G, Langbein L, Franke WW, Weitz J, Zoller M (2007). A complex of EpCAM, claudin-7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol Cancer Res 5(6): 553-567. doi: 10.1158/1541-7786.mcr-06-0384
- Gaber A, Kim SJ, Kaake RM, Bencina M, Krogan N, Sali A, Pavsic M, Lenarcic B (2018). EpCAM homo-oligomerization is not the basis for its role in cell-cell adhesion. Sci Rep 8(1): 13269. doi: 10.1038/s41598- 018-31482-7
- Guillemot JC, Naspetti M, Malergue F, Montcourrier P, Galland F, Naquet P (2001). Ep-CAM transfection in thymic epithelial cell lines triggers the formation of dynamic actin-rich protrusions involved in the organization of epithelial cell layers. Histochem Cell Biol 116(4): 371-378. doi: 10.1007/s004180100329
- Winter MJ, Nagelkerken B, Mertens AE, Rees-Bakker HA, Briaire-de Bruijn IH, Litvinov SV (2003). Expression of Ep-CAM shifts the state of cadherin-mediated adhesions from strong to weak. Exp Cell Res 285(1): 50-58. doi: 10.1016/s0014-4827(02)00045-9
- Osta WA, Chen Y, Mikhitarian K, Mitas M, Salem M, Hannun YA, Cole DJ, Gillanders WE (2004). EpCAM is overexpressed in breast cancer and is a potential target for breast cancer gene therapy. Cancer Res 64(16): 5818-5824. doi: 10.1158/0008-5472.can-04-0754
- Wu CJ, Mannan P, Lu M, Udey MC (2013). Epithelial cell adhesion molecule (EpCAM) regulates claudin dynamics and tight junctions. J Biol Chem 288(17): 12253-12268. doi: 10.1074/jbc.M113.457499
- Ladwein M, Pape UF, Schmidt DS, Schnolzer M, Fiedler S, Langbein L, Franke WW, Moldenhauer G, Zoller M (2005). The cell-cell adhesion molecule EpCAM interacts directly with the tight junction protein claudin-7. Exp Cell Res 309(2): 345-357. doi: 10.1016/j.yexcr.2005.06.013
- Lei Z, Maeda T, Tamura A, Nakamura T, Yamazaki Y, Shiratori H, Yashiro K, Tsukita S, Hamada H (2012). EpCAM contributes to formation of functional tight junction in the intestinal epithelium by recruiting claudin proteins. Dev Biol 371(2): 136-145. doi: 10.1016/j.ydbio.2012.07.005
- Huang L, Yang Y, Yang F, Liu S, Zhu Z, Lei Z, Guo J (2018). Functions of EpCAM in physiological processes and diseases (Review). Int J Mol Med 42(4): 1771-1785. doi: 10.3892/ijmm.2018.3764
- Eberlein C, Rooney C, Ross SJ, Farren M, Weir HM, Barry ST (2015). E-Cadherin and EpCAM expression by NSCLC tumor cells associate with normal fibroblast activation through a pathway initiated by integrin alphavbeta6 and maintained through TGFbeta signalling. Oncogene 34(6): 704-716. doi: 10.1038/onc.2013.600
- Munz M, Baeuerle PA, Gires O (2009). The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res 69(14): 5627- 5629. doi: 10.1158/0008-5472.can-09-0654
- Maaser K, Borlak J (2008). A genome-wide expression analysis identifies a network of EpCAM-induced cell cycle regulators. Br J Cancer 99(10): 1635-1643. doi: 10.1038/sj.bjc.6604725
- Litvinov SV, van Driel W, van Rhijn CM, Bakker HA, van Krieken H, Fleuren GJ, Warnaar SO (1996). Expression of Ep-CAM in cervical squamous epithelia correlates with an increased proliferation and the disappearance of markers for terminal differentiation. Am J Pathol 148(3): 865-875. PMID: 8774141
- Imrich S, Hachmeister M, Gires O (2012). EpCAM and its potential role in tumor-initiating cells. Cell Adh Migr 6(1): 30-38. doi: 10.4161/cam.18953
- Chaves-Perez A, Mack B, Maetzel D, Kremling H, Eggert C, Harreus U, Gires O (2013). EpCAM regulates cell cycle progression via control of cyclin D1 expression. Oncogene 32(5): 641-650. doi: 10.1038/onc.2012.75
- Liang KH, Tso HC, Hung SH, Kuan, II, Lai JK, Ke FY, Chuang YT, Liu IJ, Wang YP, Chen RH, Wu HC (2018). Extracellular domain of EpCAM enhances tumor progression through EGFR signaling in colon cancer cells. Cancer Lett 433: 165-175. doi: 10.1016/j.canlet.2018.06.040
- Ng VY, Ang SN, Chan JX, Choo AB (2010). Characterization of epithelial cell adhesion molecule as a surface marker on undifferentiated human embryonic stem cells. Stem Cells 28(1): 29- 35. doi: 10.1002/stem.221
- Sarrach S, Huang Y, Niedermeyer S, Hachmeister M, Fischer L, Gille S, Pan M, Mack B, Kranz G, Libl D, Merl-Pham J, Hauck SM, Paoluzzi Tomada E, Kieslinger M, Jeremias I, Scialdone A, Gires O (2018). Spatiotemporal patterning of EpCAM is important for murine embryonic endo- and mesodermal differentiation. Sci Rep 8(1): 1801. doi: 10.1038/s41598-018-20131-8
- Cirulli V, Crisa L, Beattie GM, Mally MI, Lopez AD, Fannon A, Ptasznik A, Inverardi L, Ricordi C, Deerinck T, Ellisman M, Reisfeld RA, Hayek A (1998). KSA antigen Ep-CAM mediates cell-cell adhesion of pancreatic epithelial cells: morphoregulatory roles in pancreatic islet development. J Cell Biol 140(6): 1519-1534. doi: 10.1083/jcb.140.6.1519
- de Boer CJ, van Krieken JH, Janssen-van Rhijn CM, Litvinov SV (1999). Expression of Ep-CAM in normal, regenerating, metaplastic, and neoplastic liver. J Pathol 188(2): 201-206. doi: 10.1002/(sici)1096- 9896(199906)188:2<201::aid-path339>3.0.co;2-8
- Hanna JH, Saha K, Jaenisch R (2010). Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell 143(4): 508-525. doi: 10.1016/j.cell.2010.10.008
- Lu TY, Lu RM, Liao MY, Yu J, Chung CH, Kao CF, Wu HC (2010). Epithelial cell adhesion molecule regulation is associated with the maintenance of the undifferentiated phenotype of human embryonic stem cells. J Biol Chem 285(12): 8719-8732. doi: 10.1074/jbc.M109.077081
- Kuan, II, Liang KH, Wang YP, Kuo TW, Meir YJ, Wu SC, Yang SC, Lu J, Wu HC (2017). EpEX/EpCAM and Oct4 or Klf4 alone are sufficient to generate induced pluripotent stem cells through STAT3 and HIF2alpha. Sci Rep 7: 41852. doi: 10.1038/srep41852
- Gires O, Klein CA, Baeuerle PA (2009). On the abundance of EpCAM on cancer stem cells. Nat Rev Cancer 9(2): 143. doi: 10.1038/nrc2499-c1
- Sato R, Semba T, Saya H, Arima Y (2016). Concise Review: Stem Cells and Epithelial-Mesenchymal Transition in Cancer: Biological Implications and Therapeutic Targets. Stem Cells 34(8): 1997-2007. doi: 10.1002/stem.2406
- Wahl GM, Spike BT (2017). Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ Breast Cancer 3: 14. doi: 10.1038/s41523-017-0012-z
- Lin CW, Liao MY, Lin WW, Wang YP, Lu TY, Wu HC (2012). Epithelial cell adhesion molecule regulates tumor initiation and tumorigenesis via activating reprogramming factors and epithelial- mesenchymal transition gene expression in colon cancer. J Biol Chem 287(47): 39449-39459. doi: 10.1074/jbc.M112.386235
- Nieto MA, Huang RY, Jackson RA, Thiery JP (2016). EMT: 2016. Cell 166(1): 21-45. doi: 10.1016/j.cell.2016.06.028
- Jojovic M, Adam E, Zangemeister-Wittke U, Schumacher U (1998). Epithelial glycoprotein-2 expression is subject to regulatory processes in epithelial-mesenchymal transitions during metastases: an investigation of human cancers transplanted into severe combined immunodeficient mice. Histochem J 30(10): 723-729. PMID: 9873999
- Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC, Manjili MH, Radisky DC, Ferrone S, Knutson KL (2009). Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res 69(7): 2887-2895. doi: 10.1158/0008-5472.can-08-3343
- Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA, Mani SA (2010). Core epithelial-to- mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 107(35): 15449-15454. doi: 10.1073/pnas.1004900107
- Hyun KA, Koo GB, Han H, Sohn J, Choi W, Kim SI, Jung HI, Kim YS (2016). Epithelial-to-mesenchymal transition leads to loss of EpCAM and different physical properties in circulating tumor cells from metastatic breast cancer. Oncotarget 7(17): 24677-24687. doi: 10.18632/oncotarget.8250
- Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, Nilsson MB, Gudikote J, Tran H, Cardnell RJ, Bearss DJ, Warner SL, Foulks JM, Kanner SB, Gandhi V, Krett N, Rosen ST, Kim ES, Herbst RS, Blumenschein GR, Lee JJ, Lippman SM, Ang KK, Mills GB, Hong WK, Weinstein JN, et al. (2013). An epithelial- mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 19(1): 279-290. doi: 10.1158/1078-0432.ccr-12-1558
- Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, Weinberg RA (2015). Distinct EMT programs control normal mammary stem cells and tumor-initiating cells. Nature 525(7568): 256-260. doi: 10.1038/nature14897
- Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, Deschler DG, Varvares MA, Mylvaganam R, Rozenblatt-Rosen O, Rocco JW, Faquin WC, Lin DT, Regev A, Bernstein BE (2017). Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171(7): 1611-1624.e1624. doi: 10.1016/j.cell.2017.10.044
- Sankpal NV, Fleming TP, Sharma PK, Wiedner HJ, Gillanders WE (2017). A double-negative feedback loop between EpCAM and ERK contributes to the regulation of epithelial-mesenchymal transition in cancer. Oncogene 36(26): 3706-3717. doi: 10.1038/onc.2016.504
- Driemel C, Kremling H, Schumacher S, Will D, Wolters J, Lindenlauf N, Mack B, Baldus SA, Hoya V, Pietsch JM, Panagiotidou P, Raba K, Vay C, Vallbohmer D, Harreus U, Knoefel WT, Stoecklein NH, Gires O (2014). Context-dependent adaption of EpCAM expression in early systemic esophageal cancer. Oncogene 33(41): 4904-4915. doi: 10.1038/onc.2013.441
- Vannier C, Mock K, Brabletz T, Driever W (2013). Zeb1 regulates E- cadherin and Epcam (epithelial cell adhesion molecule) expression to control cell behavior in early zebrafish development. J Biol Chem 288(26): 18643-18659. doi: 10.1074/jbc.M113.467787
- Gao J, Yan Q, Wang J, Liu S, Yang X (2015). Epithelial-to- mesenchymal transition induced by TGF-beta1 is mediated by AP1- dependent EpCAM expression in MCF-7 cells. J Cell Physiol 230(4): 775-782. doi: 10.1002/jcp.24802
- Maghzal N, Vogt E, Reintsch W, Fraser JS, Fagotto F (2010). The tumor-associated EpCAM regulates morphogenetic movements through intracellular signaling. J Cell Biol 191(3): 645-659. doi: 10.1083/jcb.201004074
- Gaiser MR, Lammermann T, Feng X, Igyarto BZ, Kaplan DH, Tessarollo L, Germain RN, Udey MC (2012). Cancer-associated epithelial cell adhesion molecule (EpCAM; CD326) enables epidermal Langerhans cell motility and migration in vivo. Proc Natl Acad Sci U S A 109(15): E889-897. doi: 10.1073/pnas.1117674109
- Sankpal NV, Mayfield JD, Willman MW, Fleming TP, Gillanders WE (2011). Activator protein 1 (AP-1) contributes to EpCAM-dependent breast cancer invasion. Breast Cancer Res 13(6): R124. doi: 10.1186/bcr3070
- Sankpal NV, Fleming TP, Gillanders WE (2013). EpCAM modulates NF-kappaB signaling and interleukin-8 expression in breast cancer. Mol Cancer Res 11(4): 418-426. doi: 10.1158/1541-7786.mcr-12-0518
- Riethdorf S, O’Flaherty L, Hille C, Pantel K (2018). Clinical applications of the CellSearch platform in cancer patients. Adv Drug Deliv Rev 125: 102-121. doi: 10.1016/j.addr.2018.01.011
- Andree KC, van Dalum G, Terstappen LW (2016). Challenges in circulating tumor cell detection by the CellSearch system. Mol Oncol 10(3): 395-407. doi: 10.1016/j.molonc.2015.12.002
- Bidard FC, Michiels S, Riethdorf S, Mueller V, Esserman LJ, Lucci A, Naume B, Horiguchi J, Gisbert-Criado R, Sleijfer S, Toi M, Garcia- Saenz JA, Hartkopf A, Generali D, Rothe F, Smerage J, Muinelo-Romay L, Stebbing J, Viens P, Magbanua MJM, Hall CS, Engebraaten O, Takata D, Vidal-Martinez J, Onstenk W, Fujisawa N, Diaz-Rubio E, Taran FA, Cappelletti MR, Ignatiadis M, et al. (2018). Circulating Tumor Cells in Breast Cancer Patients Treated by Neoadjuvant Chemotherapy: A Meta-analysis. J Natl Cancer Inst 110(6): 560-567. doi: 10.1093/jnci/djy018
- Allard WJ, Terstappen LW (2015). CCR 20th Anniversary Commentary: Paving the Way for Circulating Tumor Cells. Clin Cancer Res 21(13): 2883-2885. doi: 10.1158/1078-0432.ccr-14-2559
- Antolovic D, Galindo L, Carstens A, Rahbari N, Buchler MW, Weitz J, Koch M (2010). Heterogeneous detection of circulating tumor cells in patients with colorectal cancer by immunomagnetic enrichment using different EpCAM-specific antibodies. BMC Biotechnol 10: 35. doi: 10.1186/1472-6750-10-35
- Swennenhuis JF, van Dalum G, Zeune LL, Terstappen LW (2016). Improving the CellSearch(R) system. Expert Rev Mol Diagn 16(12): 1291-1305. doi: 10.1080/14737159.2016.1255144
- Rao CG, Chianese D, Doyle GV, Miller MC, Russell T, Sanders RA, Jr., Terstappen LW (2005). Expression of epithelial cell adhesion molecule in carcinoma cells present in blood and primary and metastatic tumors. Int J Oncol 27(1): 49-57. doi: 10.3892/ijo.27.1.49
- Fehm T, Muller V, Aktas B, Janni W, Schneeweiss A, Stickeler E, Lattrich C, Lohberg CR, Solomayer E, Rack B, Riethdorf S, Klein C, Schindlbeck C, Brocker K, Kasimir-Bauer S, Wallwiener D, Pantel K (2010). HER2 status of circulating tumor cells in patients with metastatic breast cancer: a prospective, multicenter trial. Breast Cancer Res Treat 124(2): 403-412. doi: 10.1007/s10549-010-1163-x
- Sieuwerts AM, Kraan J, Bolt J, van der Spoel P, Elstrodt F, Schutte M, Martens JW, Gratama JW, Sleijfer S, Foekens JA (2009). Anti- epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J Natl Cancer Inst 101(1): 61-66. doi: 10.1093/jnci/djn419
- Gervasoni A, Sandri MT, Nascimbeni R, Zorzino L, Cassatella MC, Baglioni L, Panigara S, Gervasi M, Di Lorenzo D, Parolini O (2011). Comparison of three distinct methods for the detection of circulating tumor cells in colorectal cancer patients. Oncol Rep 25(6): 1669-1703. doi: 10.3892/or.2011.1231
- Krebs MG, Hou JM, Sloane R, Lancashire L, Priest L, Nonaka D, Ward TH, Backen A, Clack G, Hughes A, Ranson M, Blackhall FH, Dive C (2012). Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and -independent approaches. J Thorac Oncol 7(2): 306-315. doi: 10.1097/JTO.0b013e31823c5c16
- Gorges TM, Tinhofer I, Drosch M, Rose L, Zollner TM, Krahn T, von Ahsen O (2012). Circulating tumor cells escape from EpCAM- based detection due to epithelial-to-mesenchymal transition. BMC Cancer 12: 178. doi: 10.1186/1471-2407-12-178
- Alix-Panabieres C, Mader S, Pantel K (2017). Epithelial- mesenchymal plasticity in circulating tumor cells. J Mol Med 95(2): 133-142. doi: 10.1007/s00109-016-1500-6
- Werner S, Stenzl A, Pantel K, Todenhofer T (2017). Expression of Epithelial Mesenchymal Transition and Cancer Stem Cell Markers in Circulating Tumor Cells. Adv Exp Med Biol 994(205-228. doi: 10.1007/978-3-319-55947-6_11
- Kasimir-Bauer S, Hoffmann O, Wallwiener D, Kimmig R, Fehm T (2012). Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res 14(1): R15. doi: 10.1186/bcr3099
- Kallergi G, Agelaki S, Kalykaki A, Stournaras C, Mavroudis D, Georgoulias V (2008). Phosphorylated EGFR and PI3K/Akt signaling kinases are expressed in circulating tumor cells of breast cancer patients. Breast Cancer Res 10(5): R80. doi: 10.1186/bcr2149
- Yokobori T, Iinuma H, Shimamura T, Imoto S, Sugimachi K, Ishii H, Iwatsuki M, Ota D, Ohkuma M, Iwaya T, Nishida N, Kogo R, Sudo T, Tanaka F, Shibata K, Toh H, Sato T, Barnard GF, Fukagawa T, Yamamoto S, Nakanishi H, Sasaki S, Miyano S, Watanabe T, Kuwano H, Mimori K, Pantel K, Mori M (2013). Plastin3 is a novel marker for circulating tumor cells undergoing the epithelial-mesenchymal transition and is associated with colorectal cancer prognosis. Cancer Res 73(7): 2059-2069. doi: 10.1158/0008-5472.can-12-0326
- Agerbaek MO, Bang-Christensen SR, Yang MH, Clausen TM, Pereira MA, Sharma S, Ditlev SB, Nielsen MA, Choudhary S, Gustavsson T, Sorensen PH, Meyer T, Propper D, Shamash J, Theander TG, Aicher A, Daugaard M, Heeschen C, Salanti A (2018). The VAR2CSA malaria protein efficiently retrieves circulating tumor cells in an EpCAM-independent manner. Nat Commun 9(1): 3279. doi: 10.1038/s41467-018-05793-2
- Lamouille S, Derynck R (2007). Cell size and invasion in TGF-beta- induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 178(3): 437-451. doi: 10.1083/jcb.200611146
- de Wit S, van Dalum G, Lenferink AT, Tibbe AG, Hiltermann TJ, Groen HJ, van Rijn CJ, Terstappen LW (2015). The detection of EpCAM(+) and EpCAM(-) circulating tumor cells. Sci Rep 5: 12270. doi: 10.1038/srep12270
- de Wit S, Manicone M, Rossi E, Lampignano R, Yang L, Zill B, Rengel-Puertas A, Ouhlen M, Crespo M, Berghuis AMS, Andree KC, Vidotto R, Trapp EK, Tzschaschel M, Colomba E, Fowler G, Flohr P, Rescigno P, Fontes MS, Zamarchi R, Fehm T, Neubauer H, Rack B, Alunni-Fabbroni M, Farace F, De Bono J, MJ IJ, Terstappen L (2018). EpCAM(high) and EpCAM(low) circulating tumor cells in metastatic prostate and breast cancer patients. Oncotarget 9(86): 35705-35716. doi: 10.18632/oncotarget.26298
- Lindsay CR, Faugeroux V, Michiels S, Pailler E, Facchinetti F, Ou D, Bluthgen MV, Pannet C, Ngo-Camus M, Bescher G, Caramella C, Billiot F, Remon J, Planchard D, Soria JC, Besse B, Farace F (2017). A prospective examination of circulating tumor cell profiles in non- small-cell lung cancer molecular subgroups. Ann Oncol 28(7): 1523- 1531. doi: 10.1093/annonc/mdx156
- Sun YF, Guo W, Xu Y, Shi YH, Gong ZJ, Ji Y, Du M, Zhang X, Hu B, Huang A, Chen GG, Lai PBS, Cao Y, Qiu SJ, Zhou J, Yang XR, Fan J (2018). Circulating Tumor Cells from Different Vascular Sites Exhibit Spatial Heterogeneity in Epithelial and Mesenchymal Composition and Distinct Clinical Significance in Hepatocellular Carcinoma. Clin Cancer Res 24(3): 547-559. doi: 10.1158/1078-0432.ccr-17-1063
- Kallergi G, Papadaki MA, Politaki E, Mavroudis D, Georgoulias V, Agelaki S (2011). Epithelial to mesenchymal transition markers expressed in circulating tumor cells of early and metastatic breast cancer patients. Breast Cancer Res 13(3): R59. doi: 10.1186/bcr2896
- Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, Brachtel E, Sgroi D, Baselga J, Ramaswamy S, Toner M, Haber DA, Maheswaran S (2013). Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339(6119): 580-584. doi: 10.1126/science.1228522
- Xu R, Rai A, Chen M, Suwakulsiri W, Greening DW, Simpson RJ (2018). Extracellular vesicles in cancer – implications for future improvements in cancer care. Nat Rev Clin Oncol 15(10): 617-638. doi: 10.1038/s41571-018-0036-9
- Meehan B, Rak J, Di Vizio D (2016). Oncosomes – large and small: what are they, where they came from? J Extracell Vesicles 5: 33109. doi: 10.3402/jev.v5.33109
- Minciacchi VR, Freeman MR, Di Vizio D (2015). Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol 40: 41-51. doi: 10.1016/j.semcdb.2015.02.010
- Andreu Z, Yanez-Mo M (2014). Tetraspanins in extracellular vesicle formation and function. Front Immunol 5: 442. doi: 10.3389/fimmu.2014.00442
- Le Naour F, Andre M, Greco C, Billard M, Sordat B, Emile JF, Lanza F, Boucheix C, Rubinstein E (2006). Profiling of the tetraspanin web of human colon cancer cells. Mol Cell Proteomics 5(5): 845-857. doi: 10.1074/mcp.M500330-MCP200
- Jiang L, Shen Y, Guo D, Yang D, Liu J, Fei X, Yang Y, Zhang B, Lin Z, Yang F, Wang X, Wang K, Wang J, Cai Z (2016). EpCAM-dependent extracellular vesicles from intestinal epithelial cells maintain intestinal tract immune balance. Nat Commun 7: 13045. doi: 10.1038/ncomms13045
- Tauro BJ, Greening DW, Mathias RA, Mathivanan S, Ji H, Simpson RJ (2013). Two distinct populations of exosomes are released from LIM1863 colon carcinoma cell-derived organoids. Mol Cell Proteomics 12(3): 587-598. doi: 10.1074/mcp.M112.021303
- Im H, Shao H, Park YI, Peterson VM, Castro CM, Weissleder R, Lee H (2014). Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat Biotechnol 32(5): 490-495. doi: 10.1038/nbt.2886
- Castillo J, Bernard V, San Lucas FA, Allenson K, Capello M, Kim DU, Gascoyne P, Mulu FC, Stephens BM, Huang J, Wang H, Momin AA, Jacamo RO, Katz M, Wolff R, Javle M, Varadhachary G, Wistuba, II, Hanash S, Maitra A, Alvarez H (2018). Surfaceome profiling enables isolation of cancer-specific exosomal cargo in liquid biopsies from pancreatic cancer patients. Ann Oncol 29(1): 223-229. doi: 10.1093/annonc/mdx542
- Karabulut S, Tas F, Tastekin D, Karabulut M, Yasasever CT, Ciftci R, Guveli M, Fayda M, Vatansever S, Serilmez M, Disci R, Aydiner A (2014). The diagnostic, predictive, and prognostic role of serum epithelial cell adhesion molecule (EpCAM) and vascular cell adhesion molecule-1 (VCAM-1) levels in breast cancer. Tumor Biol 35(9): 8849- 8860. doi: 10.1007/s13277-014-2151-2
- Kimura H, Kato H, Faried A, Sohda M, Nakajima M, Fukai Y, Miyazaki T, Masuda N, Fukuchi M, Kuwano H (2007). Prognostic significance of EpCAM expression in human esophageal cancer. Int J Oncol 30(1): 171-179. doi: 10.3892/ijo.30.1.171
- Gebauer F, Struck L, Tachezy M, Vashist Y, Wicklein D, Schumacher U, Izbicki JR, Bockhorn M (2014). Serum EpCAM expression in pancreatic cancer. Anticancer Res 34(9): 4741-4746. PMID: 25202052
- Karabulut S, Tastekin D, Ciftci R, Tambas M, Dagoglu N, Guveli ME, Tas F, Duranyildiz D, Aydiner A (2014). Clinical significance of serum epithelial cell adhesion molecule (EPCAM) and vascular cell adhesion molecule-1 (VCAM-1) levels in lung cancer. J Clin Oncol 32(15_suppl): e22192-e22192. doi: 10.1200/jco.2014.32.15_suppl.e22192
- Abe H, Kuroki M, Imakiire T, Yamauchi Y, Yamada H, Arakawa F, Kuroki M (2002). Preparation of recombinant MK-1/Ep-CAM and establishment of an ELISA system for determining soluble MK-1/Ep- CAM levels in sera of cancer patients. J Immunol Methods 270(2): 227-233. doi: 10.1016/s0022-1759(02)00332-0
- Schmetzer O, Moldenhauer G, Nicolaou A, Schlag P, Riesenberg R, Pezzutto A (2012). Detection of circulating tumor-associated antigen depends on the domains recognized by the monoclonal antibodies used: N-terminal trimmed EpCAM-levels are much higher than untrimmed forms. Immunol Lett 143(2): 184-192. doi: 10.1016/j.imlet.2012.02.004
- Petsch S, Gires O, Ruttinger D, Denzel S, Lippold S, Baeuerle PA, Wolf A (2011). Concentrations of EpCAM ectodomain as found in sera of cancer patients do not significantly impact redirected lysis and T- cell activation by EpCAM/CD3-bispecific BiTE antibody MT110. mAbs 3(1): 31-37. doi: 10.4161/mabs.3.1.14193
ACKNOWLEDGMENTS
K.P. has received research funding from the Deutsche Forschungsgemeinschaft (DFG) SPP2084 µBone, Deutsche Krebshilfe “Translationale Onkologie” n°70112507, EU TRANSCAN-199 Grant PROLIPSY, ERC PoC Grant CTCapture_2.0 n°7544533 H2020 and the EU/IMI consortium CANCER-ID under grant agreement n° 115749, European Union’s Seventh Framework Programme (FP7/2007e2013). L.K. was fi-nancially supported by ITMO Cancer “Plan Cancer 2014-2019 “and received funding from Fondation de France. S.W. has received funding by Wilhelm Sander-Stiftung 2015.148.2 and Erich und Gertrud Roggenbuck-Stiftung.
COPYRIGHT
© 2019

Biology and clinical relevance of EpCAM by Keller et al. is licensed under a Creative Commons Attribution 4.0 International License.


