Reviews:
Cell Stress, Vol. 3, No. 5, pp. 141 - 161; doi: 10.15698/cst2019.05.186
Recent progress in the role of autophagy in neurological diseases

1 State Key Laboratory of Respiratory Disease, School of Basic Medical Sciences, Guangzhou Medical University; Affiliated Cancer Hospital of Guangzhou Medical University, Guangzhou 511436, China.
2 Institute of Neurology, Guangdong Key Laboratory of Age-Related Cardiac-Cerebral Vascular Disease, Affiliated Hospital of Guangdong Medical College, Zhanjiang, Guangdong, China.
3 Department of Biochemistry and Molecular Biology, GMU-GIBH Joint School of Life Sciences, Guangzhou Medical University, Guangzhou 511436, People’s Republic of China.
# These authors contributed equally to this work.
Keywords: autophagy, neuro-degenerative diseases, mTOR, Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, Amyotrophic lateral sclerosis.
Abbreviatons:
Aβ – amyloid beta,
AD – Alzheimer’s disease,
ALS – amyotrophic lateral sclerosis,
AMPK – AMP-activated protein kinase,
APP – amyloid precursor protein,
ATG – autophagy related,
AV – autophagic vacuole,
C9ORF72 – Chromosome 9 open reading frame 72,
CMA – chaperone-mediated autophagy,
CTF – C-terminal fragment,
ESCRT – endosomal sorting complexes required for transport,
FTD – frontotemporal dementia,
HD – Huntington’s disease,
HTT – huntingtin,
mHTT – mutant HTT,
mTOR – mammalian target of rapamycin,
MVB – multi-vesicular body,
PD – Parkinson’s disease,
PI3K – phosphatidyl inositol 3-kinase, polyQ – poly-glutamine,
VCP – valosin-containing protein.
Received originally: 05/01/2019 Received in revised form: 27/03/2019
Accepted: 10/04/2019
Published: 29/04/2019
Correspondence:
Dr. Du Feng, State Key Laboratory of Respiratory Disease, School of Basic Medical Sciences, Guangzhou Medical University; Affiliated Cancer Hospital of Guangzhou Medical University, Guangzhou 511436, China feng_du@foxmail.com
Conflict of interest statement: The authors declare that they have no conflict of inter-ests.
Please cite this article as: Tian Meng, Shiyin Lin, Haixia Zhuang, Haofeng Huang, Zhengjie He, Yongquan Hu, Qing Gong and Du Feng (2019). Re-cent progress in the role of autophagy in neurological diseases. Cell Stress 3(5): 141-161. doi: 10.15698/cst2019.05.186
Abstract
Autophagy (here refers to macroautophagy) is a catabolic pathway by which large protein aggregates and damaged organelles are first sequestered into a double-membraned structure called autophagosome and then delivered to lysosome for destruction. Recently, tremendous progress has been made to elucidate the molecular mechanism and functions of this essential cellular metabolic process. In addition to being either a rubbish clearing system or a cellular surviving program in response to different stresses, autophagy plays important roles in a large number of pathophysiological conditions, such as cancer, diabetes, and especially neurodegenerative disorders. Here we review recent progress in the role of autophagy in neurological diseases and discuss how dysregulation of autophagy initiation, autophagosome formation, maturation, and/or autophagosome-lysosomal fusion step contributes to the pathogenesis of these disorders in the nervous system.
AUTOPHAGY
Autophagy is an evolutionary conserved cellular process, which is characterized by the appearance of double-membrane autophagosomes sequestering portions of cellular organelles and cytoplasm and subsequently delivering them to the lysosome for degradation [1][2]. After destruction of the autophagic cargo, amino acids, nucleotides, fatty acids, sugars, the building blocks are released into the cytosol and reutilized in metabolic pathways [3]. Therefore, autophagy is crucial for maintaining cellular homeostasis as well as remodeling during normal development, and plays a critical role in overcoming adverse conditions, such as starvation and intrinsic or extrinsic cellular stresses (hypoxia, reactive oxygen species accumulation, endoplasmic reticulum stress and bacterial infections) [4]. Dysfunctions in autophagy have been associated with a variety of pathologies including cancer [5][6][7][8], neurodegenerative diseases [9][10][11][12][13], inflammatory diseases [14][15], metabolic diseases [6][16][17] and heart dysfunction [18][19][20].
–
The formation of the autophagosome is dominated by a series of autophagy-related (ATG) genes and protein complexes acting sequentially, so that autophagy is induced when needed, but otherwise maintained at a basal level. The ULK1 complex (ULK1/2–ATG13–FIP200–ATG101) is in charge of autophagy induction, the class III phosphatidylinositol 3-kinase (PI3K)/VPS34 complex (VPS34, Beclin 1, ATG14 and VSP15 form the core of this complex, while Bif, Ambra1 and UVRAG, positively regulate its activity) is in charge of autophagosome initiation, ATG12–ATG5–ATG16L1 and LC3-I/LC3-PE (LC3-II) complexes are in charge of the extension and closure of the autophagosome double membranes (Figure 1). After autophagosome maturation, its outer membrane fuses with the lysosome membrane, the inner membrane and contents are degraded by hydrolases in the lysosome, thus generating amino acids and other cellular building blocks recycled by the cell, and this process is also a quality control mechanism for cellular organelles and proteins [21][22][23].
–
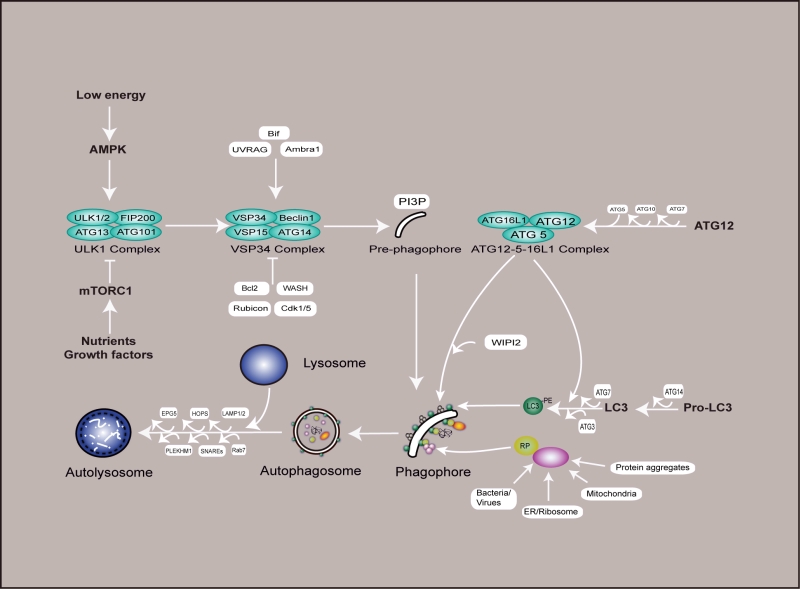 |
FIGURE 1: Schematic of the mammalian autophagy pathway. This diagram shows a simplified version of autophagy. Nutrient or growth factor deprivation and low energy are well established autophagy inducers, leading to AMPK activation and mTORC1 inhibition, which positively trigger the formation of ULK1 complex (ULK1, ULK2, ATG13, FIP200 and ATG101). This complex subsequently activates the VSP34 complex (VSP34, Beclin1, VSP15 and ATG14) to promote PI3P synthesis in pre-autophagosomal structures, thus the initiation of autophagy has been activated. PI3P specifically binds its effector WIPI2 and catalyzes two types of ubiquitination-like reactions that are in charge of the extension and closure of the autophagosome double membranes. In the first reaction, ATG12 and ATG5 are conjugated to each other in the presence of ATG7 and ATG10, and ATG16L subsequently binds to them to form the ATG12–ATG5–ATG16L1 complex. In the second reaction, LC3-I and PE are conjugated to membranes in the presence of ATG14, ATG7 and ATG3, this process is facilitated by the ATG12–ATG5–ATG16L1 complex, ultimately leading to the formation of the complete autophagosome. Receptor proteins such as p62, NDP52, and NBR1 are responsible for the recognition of cytoplasmic targets (e.g., protein aggregates, damaged mitochondria, ER/ribosome, and infectious agents), and establish a bridge between LC3-II and specific ubiquitinated cargos to sustain the engulfment of a variety of substrates. In the final step of the process, the completed autophagosomes are then trafficked to fuse with lysosomes, resulting in the degradation of the vesicle contents, and this process is regulated by LAMP1/2, EPG5, HOPS, PLEKHM1 and SNAREs. AMPK - AMP-activated protein kinase; mTORC1 - mechanistic target of rapamycin complex 1; ULK1 - Unc-51-like kinase; ATG - autophagy protein; VPS34 - phosphatidylinositol 3-kinase VPS34; PI3P - phosphatidylinositol 3-phosphate; PE - phosphatidylethanolamine; RP - receptor protein. |
THE REGULATION OF AUTOPHAGY
Besides the core components mentioned above, autophagy is regulated by important nutrient-sensing pathways including the mammalian target of rapamycin complex 1 (mTORC1) and AMP-activated protein kinase (AMPK), which oppositely modulate the ULK1 complex via a series of phosphorylation events, inhibiting or activating autophagy, respectively (Figure 1).
–
Nutrients and growth factor (signaled by receptor tyrosine kinases and PI3K/AKT pathway) availability are sensed by mTORC1, which inhibits autophagy by phosphorylating both ATG13 and ULK1 at Ser 757 and by binding to the ULK1 complex, thereby inhibiting the activity of ULK1 kinase and blocking the formation of the phagophore [24][25][26][27][28]. On the other hands, low cellular energy levels activate AMPK by phosphorylation of Thr 172 and subsequently stimulates autophagy through activating ULK1 by phosphorylation of Ser 77 and Ser 317 under glucose deprivation [29] or Ser 555 under mitophagy and amino acid starvation [30]. Activation of the ULK1 complex leads to the recruitment of VPS34 to the phagophore initiation sites, thus stimulating the formation of the phosphatidylinositol 3-phosphate (PI3P) complex and the production of PI3P, which, in turn, helps to recruit ATG16L1 by WIPI2 to autophagosome formation sites [31]. Some signals act via the ATG6 orthologous Beclin 1, which promotes VPS34 activity [32][33]. While Bcl2, WASH, Rubicon, and Cdk1/5 negatively regulate the PI3P complex to suppress autophagy [34][35].
–
The ATG12–ATG5–ATG16L1 complexes and ATG8/LC3 ubiquitin-like conjugation systems are required for maintaining the phagophore expansion. Prior to this process, ATG12 is activated by ATG7 (E1-like enzyme), attached to ATG5 by ATG10 (E2-like enzyme) and then to ATG16L, and finally enters the phagophore as a ATG5-ATG12-ATG16L1 complex. LC3 also undertakes an analogous processing: it is first cleaved by ATG4, which exposes a glycine residue by cleaving the C terminus of LC3 (LC3-I), and then conjugated to the lipid phosphatidylethanolamine (PE) with the help of ATG7, ATG3 and the ATG5-ATG12-ATG16L1 complex, leading to LC3-II formation. This process is closely associated with the extension and closure of the autophagosome double membranes [36]. The recognition of cytoplasmic targets is aided by receptor proteins, such as p62/SQSTM1 (ATG19 in yeast) [37][38], OPTN [39], NDP52 [40], NBR1 [41], ALFY [42], and TRIM5 [43], which bind to ubiquitinated proteins and link them to LC3 in the phagophore. Fusion of autophagosomes with lysosomes is supported by Rab7 GTPase and the lysosomal associated membrane protein 1/2 (LAMP1/2) [44][45], where EPG5 [46][47], HOPS[48][49], PLEKHM1[50]and SNAREs[51] are also required. Autophagolysosomal contents are decomposed by lysosomal acid hydrolases, including Cathepsin B, D, and L [52][53][54].
AUTOPHAGY AND THE NERVOUS SYSTEM
The brain is often the most severely affected organ in most lysosome disorders and mutations in genes involved in autophagy pathways are usually linked to neurodegenerative disorders, indicating the heavy reliance of neurons on autophagy to maintain normal function and homeostasis.
–
In neurons, autophagic vacuoles (AVs) are generated in axons while lysosomes are concentrated mainly near the cell body, which means that there are long distances between AVs and lysosomes due to the large expanses of dendritic and axonal cytoplasm [55]. In addition, the dysfunction of cell division in neurons causes particular obstacles in preventing impaired organelles and other waste from accumulating over a life time. In contrast, mitotic cells can dilute these waste burdens by cell division [56][57].Thus, neurons are particularly vulnerable for gradually losing the ability of efficiently clearing those burdens due to aging, which eventually results in abnormal accumulation of autolysosomal substrates like ubiquitinated protein aggregates, resulting in degeneration of neurons[30][58].
–
Although those ubiquitinated substrates can be cleared through autophagy or the ubiquitin proteasome system, autophagy is the only route to degrade large impaired organelles or protein aggregates, because they are too large to go into the narrow entrance of the proteasome chamber in the proteasome pathway. This highlights the essential role of autophagy in protein degradation and recycling in the mammalian nervous system.
AUTOPHAGY IN NEURODEGENERATIVE DISEASES
Increasing evidence has confirmed the importance of autophagy in neuronal health, and a strong link between autophagy and neurodegenerative diseases has been established based on its role of clearing abnormal aggregated proteins [59]. In fact, the intra neuronal aggregated proteins, which appear in the most late-onset neurodegenerative diseases, are usually the substrates for autophagic degradation [60][61]. The vast majority of neurodegenerative diseases, including sporadic forms and familial forms, are associated with inherited genetic mutations, and the assessment of the functions of these disease-associated genes has indicated autophagic dysfunction in pathogenesis [61]. However, the contribution of autophagy dysfunction to neurodegenerative disease progression is unknown.
ALZHEIMER'S DISEASE
Alzheimer's disease (AD) is the most common neurodegenerative disease that is characterized by extensive loss of cognitive functions. The main pathological hallmarks of AD are extracellular senile plaques which are composed of aggregated β-amyloid (Aβ) and intracellular neurofibrillary tangles (NFT) which are made of aggregated hyperphosphorylated tau protein [62].
–
Aβ originates from proteolysis of the amyloid precursor protein (APP) by the sequential enzymatic actions of β-site APP-cleaving enzyme 1 (BACE1), β-secretase, and γ-secretase, a protein complex with presenilin 1 (PS1) at its catalytic core. In AD brains, a high level of APP proteins, Aβ and PS1, accumulate in AVs in swollen dystrophic neurites, and autophagy activation was elevated after Aβ stimulation or in APP/PS1 mice, a mouse model of AD, indicating that autophagy is implicated in AD pathogenesis [63][64][65].There is a complex relation between Aβ and autophagy. Aβ may be generated in AVs during autophagy, ATG7 deletion results in lower Aβ extracellular secretion and plaque formation in APP transgenic mice [66], suggesting that the activation of autophagy may further exacerbate AD pathogenesis in AD brains [65]. However, Aβ may also be degraded by autophagy, it has been reported that enhancement of autophagy can reduce Aβ levels in a number of systems [67][68][69][70].
–
Hyperphosphorylated tau accumulation into intracellular tangles is another pathological hallmark of AD, and is also found in other neuronal diseases, such as frontotemporal dementias (FTDs) [71]. Abnormal tau disrupts vesicle transport in axons by destroying the dynein-dynactin complex, raising the number of autophagosomes and leading to tau-induced toxicity in AD and FTDs [72][73]. New data suggested that autophagy is able to degrade both soluble and aggregated forms of tau. Thus, the inhibition of autophagy accelerates tau aggregation and toxicity, and in contrast, treatment with rapamycin, an autophagy activator, decreases tau pathology [74][75][76]. This could be further confirmed by the studies in transgenic mouse models, and the results indicated that autophagy activation can suppress the formation of tau pathology and subsequently ameliorate cognitive deficits [77][78][79]. Moreover, impaired lysosomal membrane integrity was also emerged in AD patients [80], and tau has been reported to perturb lysosomal permeability by binding the membrane of lysosomes both in vitro and in vivo [76][81].
–
In healthy neurons, AVs are efficiently disposed, but in AD brains the impaired clearance of AVs, not induction of autophagy itself, results in the accumulation of autophagic vacuoles [30], indicating that the adjustment regulation of the late steps of autophagy could be a possible therapeutic strategy for AD. A further study demonstrated that the enhancement of AVs is found in PS1-rich locations [82], and knock down of PS1 leads to defects in autophagosome clearance, lysosomal acidification, and lysosomal proteolytic activity. Mutations of PS1 result in similar abnormalities in the autolysosomal pathway and are associated with early-onset AD [83]. Moreover, lack of phosphorylation on Ser367 of PS1 blocks the fusion of autophagosome and lysosome, and leads to Aβ accumulation in the mouse brain by reducing β C-terminal fraction (CTF) degradation [84]. A recent study points out that phosphorylated PS1 is capable of interacting with Annexin A2 which regulates the autophagosome-autolysosome fusion by promoting the combination of Vamp8 and autophagosomal SNARE Syntaxin 17 [84]. Based on these observations, it is reasonable to propose that recovering lysosome function may enhance the clearance of protein aggregates. And this can be further confirmed by the results that the deletion of cystatin B, an inhibitor of lysosome cysteine proteases, promotes the removal of aberrant protein aggregations in lysosomes of AD mice [85].
–
Recently, new mechanistic insights proposed that autophagic pathology in AD is caused by abnormal axonal retrograde transport of AVs. Aβ oligomers can bind to dynein intermediate chain (DIC) and cause the deficiency of dynein motors, which block its function of providing motility for retrograde transport and sending AVs to lysosomes for digestion. Hence, retrograde transport of axonal autophagosomes is obstructed and leads to autophagic stress in AD neurons [86].
–
Beclin1, the key factor in autophagosome formation, has been shown to be suppressed both on mRNA and protein levels in AD brains [87][88]. A study showed that the decline of Beclin1 on protein level is caused by caspase 3 cleavage, which is activated in AD patients brains and leads to autophagy disruption [89]. Nrf2, a vital transcription factor for regulating autophagy related protein transcription [90], could stimulate autophagy by inducing autophagy receptor NDP52 and lower aggregated tau proteins in response to oxidative stress [91]. Based on these results, the levels of Beclin1 and Nrf2 are thought to be regarded as common potential markers for pathology of AD.
–
It has been identified by genetic studies that phosphatidylinositol binding clathrin assembly protein (PICALM) is involved in autophagy [92][93], and changes in the level of this protein have been found in AD patients brains [94][95]. PICALM is a clathrin adaptor protein, and is involved in endocytic trafficking by regulating endocytosis of soluble NSF attachment protein receptors (SNAREs), thus enhancing autophagy to clear tau aggregations [96]. In addition, PICALM could act as an autophagy receptor when compounded with assembly polypeptide 2 (AP2), that own the function of interacting with LC3 and targeting APP into autophagosomes [97].
–
Researchers have found that many proteins prevent or promote AD progression via autophagy pathway. Nuclear receptor binding factor 2 (NRBF2) is a component of PI3K complex and involve in the regulation of autophagy. A study discovered that NRBF2 is reduced in the hippocampus of 5XFAD mice. However, knockout of NRBF2 can increase the level and half-life of APP-CTFs, Aβ1-40 and Aβ1-42 apparently, which demonstrates that NRBF2 plays an important role in the degradation of APP-CTFs and Aβ. In the brain of 5XFAD AD mice, NRBF2 is found to interact with APP and recruit APP and APP- CTFs into autophagic structures and trigger their degradation in autolysosome. Besides, overexpression of NRBF2 decreases p62 but enhances LC3, which means that it is able to facilitates autophagy [98].
–
Transient receptor potential Mulcolipin-1 (TRPML1), which's expression is decreased in APP/PS1 transgenic mice, is involved in the initiation of autophagy by inhibition of mTOR and activation of AMPK signaling pathway. Overexpression of TRPML1 not only decreases the expression of Beclin1, LC3 and LAMP1, but further reduces cell viability and lysosomal ion concentration which have been impaired by Aβ1-42 [99].The triggering receptor expressed on myeloid cells 2 (TREM2) is an immune receptor which recruits PI3K through adaptor DAP 10 and promotes late-onset AD. Abundant LC3II and multivesicular structures with lower expression of p62 can be observed in 5XFAD with negative Trem2 expression, which shows that the deficiency of TREM2 further induces autophagy. And it is further identified that in the TREM2-deficient microglia from AD mice or human, mTOR is inhibited while AMPK is activated. Those reactions indicate that autophagy has been further induced and results in the removal of Aβ accumulated in microglia [100]. And it is also reported that deficiency of Toll-interacting protein (Tollip) disrupts endosome-lysosome fusion and promotes the accumulation of Aβ in neurons with an enhancement of p62, Parkin and the number of autophagosomes, which are involved in autophagy and mitophagy [101]. Therefore, the deficiency of TRPML1, TREM2 or Tollip in AD cases may have a positive effect to protect neurons via autophagy.
–
In addition, ErbB2 can physically dissociate Beclin-1 from the VPS34-VPS15 complex, and suppression of ErbB2 by an inhibitor promotes autophagy activation and decreases the level of βCTF and Aβ in AD models [102]. Hence, the presence of ErbB2is unbeneficial to the development of AD.
–
Recent studies have demonstrated that autophagy-related drugs or compounds, such as the mTOR inhibitor rapamycin, can rescue the cognitive deficits and remove the aggregates (such as Aβ and tau) in AD cases efficiently [74][103]. Arctigenin, which is an extract from Arctium lappa, can induce autophagy by inhibiting AKT/mTOR pathway as well as activating AMPK/Raptor pathway, and then enhance Aβ clearance in cell and mouse models of AD [104]. The natural polyphenol resveratrol controls Aβ metabolism and mediates the anti-amyloidogenic effect through activating AMPK pathway, subsequently triggering the autophagic degradation of Aβ [105]. Functionalized single walled carbon nanotubes (SWNT) were found to restore normal autophagy by repairing aberrant activation of mTOR pathway and deficit in lysosomal proteolysis, which shows a novel neuroprotective approach in AD therapy [106]. Additionally, GTM-1 (a novel autophagy inducer) [107], latrepirdine (a pro-neurogenic, antihistaminic compound) [108], GSK-3 inhibitor (such as L803-mts) [109, 0], trehalose (a natural disaccharide) [78][79], temsirolimus (a compound for renal cell carcinoma treatment) [111] and nilotinib (a drug for adult chronic myelogenous leukemia treatment) [112] also exert functions of autophagy induction and antagonism against Aβ toxicity in AD cases. Many active ingredients extracted from traditional Chinese herbs, like DDPU [113] (a ginsenoside derivative), berberine [114] (an isoquinoline alkaloid isolated from the coptidis Rhizoma), DNLA [115] (an active ingredientextracted from Dendrobium nobile Lindl) also have therapeutic effects in AD models. The autophagy targets of these compounds are listed in Table 1.
–
Table 1. Autophagy-related potential drugs for the treatment of neurodegenerative diseases. |
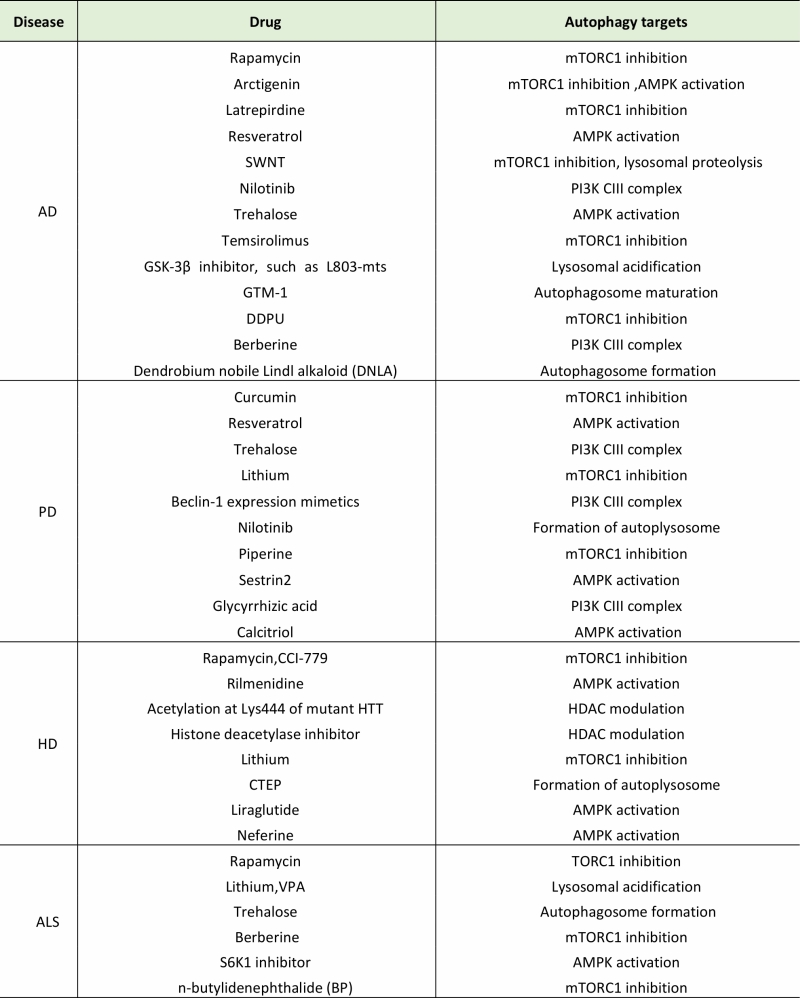 |
PARKINSON'S DISEASE
Parkinson's disease (PD) is the second most prevalent neurodegenerative disease after AD, and it is characterized by selective loss of dopamine neurons in the substantia nigra (SN) and the presence of Lewy bodies, which are composed of α-synuclein and poly-ubiquitinated proteins [116]. In addition to motor syndromes such as resting tremor and muscular rigidity, PD patients also suffer from non-motor psychological and somatic symptoms [117], these influence human normal life seriously, and the main triggers of this disease are a combination of genetic predisposition and environmental factors.
–
In brains of PD patients, aberrant lysosomes and aggregated autophagosomes were observed in neurons [118], indicating a relationship between autophagy and PD. One of the pathological hallmarks of PD is accumulation of Lewy bodies, main components of which are misfolded and aggregated α-synuclein [119, 0]. The pathogenic role of autophagy in PD was revealed by increasing levels of α-synuclein when lysosomes are inhibited, and misfolded α-synuclein oligomers can be removed by different catabolic pathways including macroautophagy and chaperone-mediated autophagy (CMA) with different pathological situations, while α-synuclein monomers are also degraded by the proteasome [121][122], suggesting a close link between α-synuclein degradation and autophagy. Furthermore, both over-expression of wild-type α-synuclein or A30P and A53T mutations of α-synuclein can inhibit autophagy [123][124], and up-regulation of transcription factor EB (TFEB), a key autophagy modulator [125], could alleviate lysosomal damage by promoting its biogenesis, thus relieving α-synuclein associated pathology of neurodegenerative diseases [126][127].
–
Emerging results have suggested that aberrant autophagy is one of the underlying mechanisms for PD, and this can be proved by evidences that several genetic mutations are linked to autophagy in hereditary forms of PD. In autosomal dominant PD, mutations in vacuolar protein sorting-associated protein 35 (VPS35) and leucine rich repeat kinase 2 (LRRK2) are mainly present. VPS35 is a retromer complex component, which recruits the actin nucleation-promoting WASP and Scar homolog (WASH) complex to endosomes. D620N mutation of VPS35 perturbs this recruitment and causes the mislocalization of mATG9 and defect of autophagosome formation [128]. LRRK2 exhibits pleiotropic functions, recent evidence raises the possibility that the toxic actions of LRRK2 are mediated by α-synuclein [129]. Shortened neurites and autophagosomes aggregation could be observed in differentiated SH-SY5Y cells expressing G2019S mutation of LRRK2 [130], which could cooperate with α-synuclein and cause age-related deficits of autophagy in a C. elegans model [131]. In the meantime, LRRK2 is able to recruit the PI3K III complex and Rubicon to the phagosome which inhibit the maturation of the phagosome [132]. Besides, mutations in SNCA (encoding α-synuclein), CHCHD2 (encoding a mitochondrial protein) and DNAJC13 (encoding a chaperon REM-8 involved in protein trafficking) are also related with autosomal dominant PD [117].
–
In autosomal recessive forms of PD, mutations in Parkin RBR E3 ubiquitin protein ligase (Parkin) [133] and PTEN induced putative kinase 1 (Pink1) [134] are the main pathogenic factors, accounting for 50% of familial cases in Europe [135]. A deficit in striatal synaptic plasticity and evoked dopamine release response was found in the striatum of mice where Parkin was deleted [136], and impaired activity of mitochondria was also observed in striatal neuron [137]. Similarly, deletion of Pink1 also leads to impaired respiration in striatal mitochondria and enhances sensitivity to oxidative stress in the cerebral cortex of mice [138]. Indeed, these two proteins act in the same way by selectively degrading damaged mitochondria to promote mitophagy [139][140]. In this process, the proteasome-mediated degradation of Pink1 is stalled on damaged mitochondria, the accumulated Pink1 subsequently phosphorylates ubiquitin and recruits Parkin on the outer membrane of these mitochondria and results in their sequestration into autophagosomes. In the meantime, some outer membrane proteins are ubiquitinated by activated Parkin, and subsequently phosphorylated by Pink1, these linkages elicit a positive feedback involving more ubiquitinated proteins of mitochondria [139][141][142][143]. Hence, a defect in mitophagy may be the cause in Pink1 or Parkin-positive familial forms of PD due to the accumulation of damaged mitochondria and excessive reactive oxygen species (ROS) production. In addition, the mutation of DJ-1 (a mitochondrial protein involved in the moderation of oxidative stress) is also related to this forms of PD, defective morphology and reduced activity are found in dopaminergic neurons derived from DJ-1 or Pink1 knockout mice [144]. In a rotenone induced PD rat model, the reduction of LAMP-2A protein, a marker of CMA, in dopamine neurons can be rescued by overexpression of DJ-1 in astrocytes, which indicates that astrocyte-specific DJ-1 overexpression has a positive effect on CMA [145]. And Fbw7βis a F-box protein which is involved in proteasomal degradation by interacting with Parkin and protects neurons from oxidative stress. A recent study has shown that 6-OHDA facilitates oxidation and the digestion of Fbw7β mainly by CMA. However, the level of Fbw7β did not change in postmortem PD brains compared to controls, thus needing further studies in vivo in PD patients [146].
–
Genome-wide association studies (GWAS) have identified a few lysosome related genes associated with PD. Mutations in the gene GBA (glucocerebrosidase β acid), encoding lysosomal hydrolase, disturb autophagosome-lysosome pathway and cause aggregation of α-synuclein [147][148]. Lysosomal ATPases are enssential for the maintenance of lysosomal pH and, therefore, the activity of lysosomal proteases. The P-type ATPase ATP13A2 is found mutated in early-onset Parkinsonism [149][150]. Mutations in ATP13A2 down-regulate degradation in lysosomes and accumulate α-synuclein protein in dopaminergic neurons [151][152]. Recent studies show that depletion of ATP13A2 causes degradation of ubiquitinated synaptotagmin 11 (SYT11) that triggers lysosome dysfunction and impaired autophagosome degradation, and these can be rescued by overexpression of SYT11 in ATP13A2 knockdown cells [153]. Another ATPase, ATP6AP2, is required for lysosomal acidification and function, depletion of it has been related to X-linked parkinsonism with spasticity [147]. Moreover, VPS13C, having a function in maintaining the normal condition of lysosome and mitochondria, is involved in autosomal recessive Parkinsonism [147][154], and the mutations of SCARB2, encoding lysosomal integral membrane protein-2 (LIMP-2), result in defects in autophagosome or lysosome function [155][156]. Other abnormalities like oxidative stress also exhibit the involvement of autophagy in PD [157]. It has been shown that oxidative stress increases autophagic cell death in dopaminergic neurons by reducing the expression of Oxi-α, which encodes a novel mTOR activator [158]. TMEM175 is a component of the lysosome proteome which is important to regulate lysosomal pH and function. A study has discovered that higher levels of phosphorylated α-synuclein aggregates LC3 and p62 when TMEM175 is depleted in rat primary hippocampal neurons, which means a high risk of PD and damaged lysosomal degradation. In addition, TMEM175 is also involved in mitophagy via influencing mitochondrial respiration and regulating energy homeostasis. Thus, abnormal autophagy and mitophagy induced by TMEM175 deficiency might play an important role in the development of PD [159].
–
As representative candidate drugs for PD, resveratrol [160] and curcumin [161] have been reported to promote the degradation of α-synuclein by AMPK-SIRT1-autophagy pathway and mTOR/p70S6K signaling pathway respectively, both of them ameliorate the neurodegenerative pathology in cell models of PD. Trehalose enhances the clearance of mutant but not wild type α-synuclein in PC12 cells by activating autophagy [162], and nilotinib reverses motor behavior deficits and loss of dopamine neurons via autophagic degradation of α-synuclein in PD models [163]. A therapy of beclin 1 injections ameliorates the pathology of synapses and dendrites in PD model mice, and reduces α-synuclein aggregates, indicating that beclin-1 expression mimetics could be a kind of potential drugs for PD treatment [164]. Besides, piperine [165], sestrin2 [166], glycyrrhizic acid [167], calcitriolare [168] also exert anti-PD pathology properties in cell or mouse models, their specific autophagy targets are listed in Table 1.
HUNTINGTON'S DISEASE
Huntington's disease (HD), an autosomal dominant neurodegenerative disease, is the most common polyglutamine disease. This kind of neurodegenerative disorder is caused by a CAG trinucleotide repeat expansion in the first exon of the huntingtin (HTT) gene which produces a mutant form of the HTT protein (mHTT) and leads to its pathogenic aggregation [169][170]. Patients with HD suffer progressive motor, cognitive and psychiatric dysfunctions, which can be manifested by ataxia, chorea, dyskinesia, depression or memory and personality disturbances [117]. The pathogenic mechanism of HD is related to interferences in the key neuronal genes transcription, disturbances in the cytoskeletal system, impairments of mitochondrial activity and alterations in the autophagy-lysosome system [171].
–
There are aberrant relations between autophagy and the onset of HD. In the postmortem brains of HD patients, altered autophagy was observed [172], and activation of autophagy by rapamycin (a mTOR inhibitor) treatment shows a neuroprotective effect and attenuates HTT toxicity in a fly model of HD [173]. Moreover, an altered expression of autophagy-related genes has been discovered in HD patients. In this aspect, the expression of genes such as LC3I, ULK2 and LAMP2 are increased in mRNA level, while the expression of EEF1A2, FKBP1A and PINK1 is down regulated [174]. A recent study showed that the V471A polymorphism in ATG7 is related to an earlier onset form of HD [175]. The exact mechanism of autophagic dysfunction in HD is poorly understood, but the inefficient degradation of autophagosomes may be the cause of their slower turnover and HTT accumulation in HD cells. This can be proved by aggregated autophagosomes observed in cellular and animal models of HD, thus dysfunction of loading into autophagosomes causes an impaired autophagic protein degradation [176].
–
Wild type HTT plays a key role in axonal transport of autophagosomes together with Huntington associated protein 1 (HAP1) [174]. Depletion of HTT in HD models results in abnormal accumulation of autophagosomes [177], and HTT also shares resemble structure with ATG11 and mTOR to join in the formation of autophagosome [56][178]. Together with the observation that overexpression of full-length HTT stimulates activation of autophagy and promotes clearance of its mutant form [179], it is tempting to speculate that wild type HTT may have extensive interactions with autophagic pathways in HD. Further studies revealed that HTT interacts with autophagy-associated proteins to influence autophagy pathway indirectly. HTT reduces the activity of mTOR by competing with mTOR in binding to ULK1, as a result, initiation of autophagy is evoked [180][181]. Additionally, it also acts as a scaffold [182] to support translocation and binding of p62, ubiquitinated proteins and LC3 to enhance autophagy activation [180]. It's reasonable to suppose that wild type HTT may regulate autophagy in different ways and that dysfunctional autophagy may also be implicated in HD cases.
–
It seems that heterozygous forms of HD are more common in the present studies, which means that patients carry a functional HTT along with mHTT [183]. mHTT displays different properties and functions compared to normal HTT due to its expanded region of glutamine residues, and the interaction between mHTT and its target proteins can be determined by the length of its poly-glutamine (polyQ) tract [183][184]. Compared with wild type HTT, mHTT seems to mediate autophagy by different ways. In addition to pathogenic aggregation of mHTT which aggravates the condition in HD, soluble forms of mHTT also represent cytotoxicity by interacting with regulators of autophagy like beclin-1, and both of the forms can be degraded by autophagy [185][186].
–
The normal functions of HTT are essential for neuronal development. Studies found that autophagosome transport is inhibited by either loss of HTT or expression of the mutant protein in striatal neurons, subsequently obstructing the fusion of autophagosome and autolysosome [187]. Further studies found that mHTT can also interact with p62 instead of wild type HTT, leading to dysfunction of p62 to recognize cargo aggregates and organelles, causing abnormal autophagy and proteasome degradation [188][189]. Besides, mHTT is able to trigger chronic stress and prolonged unfolded protein response (UPR), resulting in lower aggregate removal and inhibition of autophagy via IRE1 [190]. Recent experiments revealed that mHTT has the ability to compete with Ataxin-3 to capture Beclin 1 via its polyQ, leading to impairment of starvation-induced autophagy in neurons [184][191]. In other words, Beclin 1 can be recruited by mHTT directly, which may be a reason for unsuccessful Beclin 1-mediated long-lived protein turnover and reduction of mHTT degradation in HD cases [184][185].
–
Rhes, which is required for autophagy by interacting with Beclin 1 and facilitating disassociation between Bcl-2 and Beclin 1 [192], is invalid when interacting with mHTT which causes the impairment of autophagy initiation [192]. mTOR, another negative regulator of autophagy, is separated by mHTT which forms aggregates around mTOR, thus reducing its activity in HD and SCA7 brains [173][189][193].
–
Moreover, the activity of some enzymes also plays a role in regulating the effect of autophagy to remove mHTT aggregates. For example, up-regulation of casein kinase 2 (CK2) reduces large inclusion formation of mHTT by phosphorylating p62[194]. Down-regulation of Phosphatidylinositol-5-phosphate 4-kinase, type II γ (PIP4Kγ) enhances basal autophagy and reduces the aggregates and total amount of mHTT protein in neurons and fibroblasts respectively and rescues mHTT-induced neurodegeneration in two Drosophila HD models [195]. In mHTT-expressing neuro2A, Glycogen synthase (GS) is activated and promotes autophagy and this response is specific in neurons. Co expression of GS and mHTT can be found in HD-associated cells which restores autophagy whereas excessive autophagy is easy to cause neuronal death [196]. Overall, mHTT has multidirectional effects on the regulation of autophagy, the ratio of soluble to aggregated mutant protein may determine the toxic or protective outcome [197].
–
It is worth to mention that the classic inhibitor of mTOR, rapamycin or its analogue CCI-779, can alleviate severity of Huntington-like phenotype in behavioral experiments and facilitate the clearance of mHTT aggregates in a mouse model of HD [173], and co-treatment of rapamycin and trehalose in mice has a synergistic effect on the induction of autophagy which may accelerate the degradation of these aggregate-prone proteins efficiently [198]. Besides, lithium may be a potential drug for the treatment of PD and HD, for it has ability to remove the abnormal accumulations of mHTT and α-synuclein by inhibiting inositol monophosphatase and thus inducing autophagy [199]. Moreover, acetylation at Lys444 of mHTT [200] and upregulation of HSC70 and lamp2A [201] have been regarded as the novel therapies to remove mHTT by autophagy. Additionally, some drugs may also be useful for the treatment of HD, such as rilmenidine [202], histone deacetylase (HDAC) inhibitors [203], CTEP (a negative allosteric modulator of metabotropic glutamate receptor 5 (mGluR5)) [204], liraglutide (a GLP-1 analogue) [205], neferine (a bisbenzylisoquinoline alkaloid isolated from the lotus seed embryo of Nelumbo nucifera Gaertn) [206], their targets are listed in Table 1.
AMYOTROPHIC LATERAL SCLEROSIS
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease with the symptoms of muscle weakness, spasticity and atrophy. Selective loss of motor neurons can be observed in the brain and spinal cord of the patients [207][208]. Environmental elements such as exposure to toxic substances or heavy metals raise the risk for developing ALS. The genetic mutations such as superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43) and Chromosome 9 open reading frame 72 (C9ORF72), fused in sarcoma/translocated in lip sarcoma (FUS/TLS) resulting in accumulation of misfolded proteins have been linked to the disease [209]. Like other neurodegenerative diseases, there are sporadic and familial forms of ALS, the sporadic form is seen in the majority of known cases, while the family ones account for approximately 5%-10% [210].
–
Numerous studies try to define the molecular pathogenesis of these devastating diseases, recently it has become apparent that the autophagic/lysosomal system dysfunction is tightly associated with ALS. Indeed, aggregated autophagosomes in the cytoplasm observed in the spinal cord of sporadic ALS patients indicate that autophagy is activated [211]. Immunohistochemical analysis in a mutant SOD1 ALS mouse model (SOD1G93A) has shown the activation of autophagy [212]. Increased autophagosome formation and decreased phosphorylation of mTOR/Ser2448 are also found in motor neurons of SOD1G93A transgenic mice, indicating that autophagy dysfunction possibly underlies pathological phenomena in ALS [213].
–
SOD1 is the most common mutated gene in ALS, and the toxic gain-of-function mutations in this gene lead to its misfolding and aggregation [214]. The two studies in SOD1 mutant mice mentioned above have shown that mutant SOD1 enhanced the function of mTOR-dependent autophagy [212][213]. Besides, p62 is also increasing at the same time, which shows that autophagy fails to degrade cellular products caused by SOD1 mutations [215][216]. Furthermore, knockdown of the UPR transcription factor X-box-binding protein-1(XBP-1) in mice stimulates autophagy and promotes digestion of mutant SOD1, which can hinder the development of ALS [217]. And the heat-shock protein (HspB8) enhanced the ability to remove aggregation and mutant SOD1 by promoting autophagy in an ALS model [218]. The efficient autophagy clearance of mutant SOD1 may be beneficial for reducing motor neuron loss in ALS.
–
Many reports have revealed that a number of autophagy receptors are encoded by ALS-linked genes, p62/SQSTM1 is one of them. p62 contains SMIC, UBA and LIR domains, which can bind to SOD1, TDP-43, and LC3, respectively [219][220]. LC3 fails to recognize p62 when ALS-associated L341V mutation occurs in such cells, whereas ubiquitinated proteins still bind to it, thus causing mutant p62 and its binding protein unable to be recruited into phagophores and interrupting the autophagy-mediated degradation pathways [219][220][221].Consequently, this leads to the accumulation of mutant SOD1 and TDP-43, further accelerating the development of ALS. Furthermore, over-expression of p62 could relieve TDP-43 aggregation by autophagy or proteasome pathway in vitro [222].
–
Another autophagy receptor which is associated with ALS is optineurin (OPTN). OPTN is a ubiquitin-binding scaffold protein and take part in selective autophagy processes. Its activity is regulated by TANK-binding kinase 1 (TBK1), a protein involved in autophagy by phosphorylating p62, OPTN [223]. Inhibiting the expression of TBK1 interrupts efficient formation and maturation of autophagosomes [224][225]. Besides, OPTN also interacts with myosin VI, and autophagosome-lysosome fusion will decrease by ALS-associated mutations in the myosin VI-binding domain of OPTN, indicating OPTN is required for autophagosome trafficking [226][227]. Furthermore, OPTN and TBK1 influence mitophagy. Pink1 and Parkin can recruit TBK1 and OPTN which act as autophagic receptor to mitochondria membrane, so that they accelerate recruitment of LC3 and promote digestion of damaged mitochondria. ALS-associated mutation in OPTN and TBK1 block the closure of depolarized mitochondria and induce the accumulation of damaged mitochondria, which can break cell homeostasis, especially in neurons [225][228][229].
–
Ubiquilin2 (UBQLN2), which acts as a proteasome shuttle factor, has an ability to mark the protein with ubiquitin label for autophagy degradation, therefore it plays a crucial role in formation of autophagosome [230]. Mutations in UBQLN2 cause dominantly inherited ALS, resulting in neuron loss, cognitive deficits and shortened lifespan in mouse models [231][231]. Mutant UBQLN2 combines with polyubiquitinated proteins prior to proteasome, leading to a defect in proteasomal degradation and accumulation of misfolded proteins [233][234].
–
C9ORF72 is the most common genetic factor giving rise to ALS, and mutations in the hexanucleotide-repeat expansion of C9ORF72 gene cause disease through a number of different mechanisms [235][236]. C9ORF72 is reduced in ALS and FTD. When C9ORF72 is deleted in neurons, the accumulation of aggregated p62 and TDP-43 will occur in the cell apparently [237]. Meanwhile, decreased activity of mTOR accompanied by enlarged lysosomal compartments and enhanced autophagic flux were found in C9ORF72 depletion cells, suggesting that C9ORF72 is related to mTOR-dependent autophagy [238]. In addition, C9ORF72 forms a complex with WDR41 and SMCR8 [239][240]. This complex acts as a GDP/GTP exchange factor (GEF) to activate Rab8a and Rab39b, thus affecting the formation or maturation of autophagosomes [224][241]. In addition, this C9ORF72 complex interacts with the ULK1 complex and is required for translocation of the later one. Loss of SMCR8 leads to a similar phenotype as C9ORF72 depletion and results in defective autophagy, indicating that this interaction is required for modulating autophagy induction [242].Whereas a recent study shows a new topic that depletion of C9ORF72 is not deleterious by itself but synergizes with Ataxin-2 toxicity to impair motor neuron's function and lead to neuronal cell death, thus revealing a double-hit pathological mechanism in ALS [237].
–
An additional familial ALS gene has been reported, namely endosomal sorting complexes required for transport (ESCRT). It is required to form functional multi-vesicular bodies (MVBs) and mediates its internalization process so that most of the substances can be degraded by autophagy. ESCRT and its subunit charged multi-vesicular body protein-2B (CHMP2B) have been identified to be associated with ALS. Depletion of ESCRT or mutation in CHMP2B inhibited autophagy due to impaired autophagosome-lysosome fusion, resulting in the accumulation of ubiquitin-positive proteins and p62 [42][243]. Besides, dysfunctional MVBs in ESCRT mutated cells weaken the ability to remove TDP-43, which is the main misfolded proteins in ALS, also ensuring the connection between ESCRT and ALS [244][245].
–
Recent studies have shown that progranulin (PGRN), a secreted growth factor, and Sigma receptor-1 (SigR1), an ER chaperone, contribute to the pathogenesis of ALS and both of them own functions in neuronal survival [246][247]. The deficiency of PGRN promotes the formation of TDP-43 aggregates and inhibits autophagy in neurons [246]. The ALS-linked E102Q mutant SigR1 aggregates, co-localizes with TDP-43 in the inclusion, leading to accumulation of p62 and LC3II and obstructing autophagosome-autolysosome fusion [247].
–
Mutant Valosin-containing protein (VCP) also has been discovered in ALS patients, and it seems to regulate autophagosome removal [248]. VCP is an indispensable component to maintain the integrity and dynamics of the lysosomal network and is subsequently implicated in the maturation and fusion of autophagosome and autolysosome [248][249][250]. A study found that mutant VCP in ALS interacts with TDP-43 genetically and causes the redistribution of TDP-43 to the cytoplasm, thus probably acting as a etiology of ALS [251][252].
–
It is corroborated that rapamycin also exerts a positive effect on the therapy of ALS by a mTOR-dependent pathway, however, its function has been argued that it cannot remove the aggregates apparently in mice expressing abnormal SOD1 [253][254]. The treatment with lithium is able to alleviate the symptoms of ALS in human and animal cases by triggering autophagy through the GSK-3β pathway, and the collaboration of lithium and valproic acid (VPA) may have a better therapeutic effect on ALS [255][256]. Trehalose can upregulate the expression of ATG5, LC3 and beclin1, and subsequently promote the formation of autphagosome to delay disease onset and prolong lifespan [257]. Besides, berberine [258], p70 S6 kinase 1 (S6K1) inhibitors [259] or n-butylidenephthalide (BP) [260] are also involved in the autophagy related therapy of ALS, their targets are listed in Table 1.
CONCLUSIONS
Autophagy acts as a ubiquitous degradative pathway of large protein aggregates and damaged organelles to maintain homeostasis and function of the neurons. To date, numerous studies have shown that autophagy plays an important role in the onset and development of neurodegeneration. To sum up, abnormal proteins which give rise to neurodegenerative diseases, such as Aβ in AD, α-synuclein in PD, HTT in HD and SOD1 in ALS, will modulate autophagy in a different manner. Dysfunction in the process of autophagy pathway, such as vesicular transportation, autophagosome formation and autophagosome-autolysosome fusion, may cause the accumulation of abnormal proteins in neurons which may exacerbate the damage of neurons. Hence, the dysfunction of autophagy process is implicated in the pathology of neurodegenerative diseases. A detailed illustration of autophagic alternations in neurodegenerative diseases is shown in Figure 2.
–
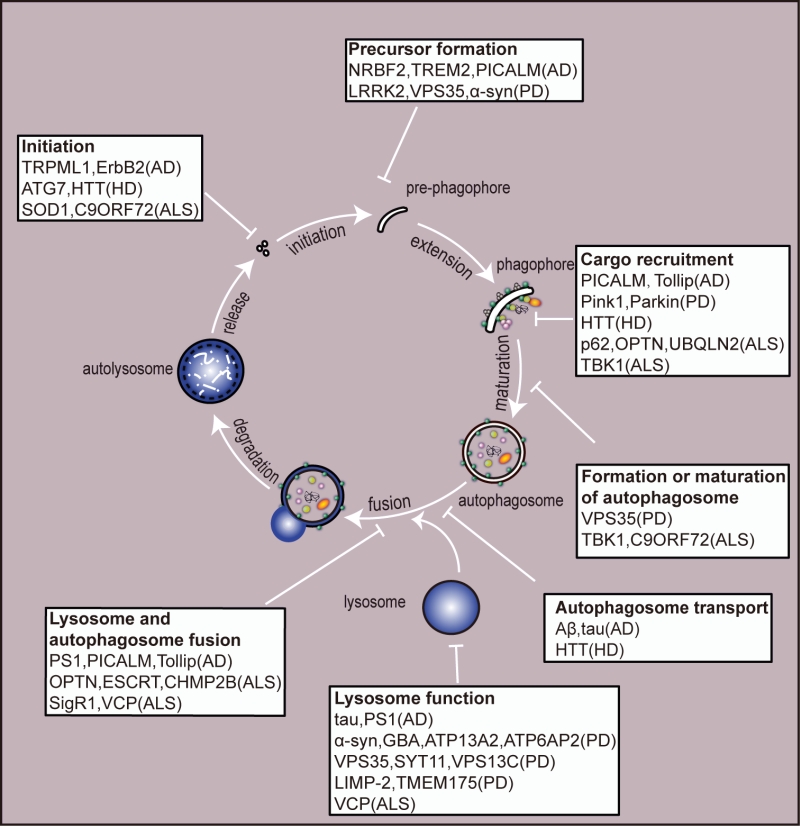 | FIGURE 2: An overview of the autophagy pathway and the site of action of disease-associated proteins. A huge number of neurodegenerative disease-related genes have been implicated in autophagy function. Mutation or deletion of these genes have been suggested to be involved in perturbation throughout the autophagic process, from initiation of autophagosome formation to degradation in the autolysosomes. Their proposed sites of action are highlighted in boxes. Please note that some disease-associated proteins act at multiple points in the process. AD - Alzheimer disease; PD - Parkinson's disease; HD - Huntington's disease; ALS - amyotrophic lateral sclerosis. |
–
It seems that the proper enhancement of autophagy may be beneficial for cell survival in neurons. Thus, autophagy will become a therapeutic target to ameliorate neurodegenerative diseases. However, the mechanism of autophagy in neurodegenerative diseases and the crosstalk between autophagy and other regulatory system, such as the immune process and inflammation, is complicated and unclear. Moreover, the specific therapeutic target of autophagy and the signal pathways involved is also undiscovered. There are still many unsolved mysteries that need further exploration.
REFERENCES
- He C, Klionsky DJ (2009). Regulation Mechanisms and Signaling Pathways of Autophagy. Annu Rev Genet 43(1): 67-93. doi: 10.1146/annurev-genet-102808-114910
- Rodolfo C, Di Bartolomeo S, Cecconi F (2016). Autophagy in stem and progenitor cells. Cell Mol Life Sci 73(3): 475-496. doi: 10.1007/s00018-015-2071-3
- Lin Y, Huang Y, Chen L, Chu P (2015). Autophagy in cancer stem/progenitor cells. Cancer Chemother Pharmacol 75(5): 879-886. doi: 10.1007/s00280-014-2634-2
- Guan J, Simon A, Prescott M, Menendez J, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A, Zhang J (2013). Autophagy in stem cells. Autophagy 9(6): 830-849. doi: 10.4161/auto.24132
- Lei Y, Zhang D, Yu J, Dong H, Zhang J, Yang S (2017). Targeting autophagy in cancer stem cells as an anticancer therapy. Cancer Lett 393: 33-39. doi: 10.1016/j.canlet.2017.02.012
- Guo JY, White E (2017). Autophagy, Metabolism, and Cancer. Cold Spring Harb Symp Quant Biol 81:73-78. doi: 10.1101/sqb.2016.81.030981
- Hua F, Shang S, Hu ZW (2017). Seeking new anti-cancer agents from autophagy-regulating natural products. J Asian Nat Prod Res 19(4): 305-313. doi: 10.1080/10286020.2017.1304385
- Jacob JA, Salmani JM, Jiang Z, Feng L, Song J, Jia X, Chen B (2017). Autophagy: An overview and its roles in cancer and obesity. Clin Chim Acta 468: 85-89. doi: 10.1016/j.cca.2017.01.028
- Chen K, Yuan R, Geng S, Zhang Y, Ran T, Kowalski E, Liu J, Li L (2017). Toll-interacting protein deficiency promotes neurodegeneration via impeding autophagy completion in high-fat diet-fed ApoE−/− mouse model. Brain Behav Immun 59: 200-210. doi: 10.1016/j.bbi.2016.10.002
- Hwang CJ, Kim YE, Son DJ, Park MH, Choi DY, Park PH, Hellstrom M, Han SB, Oh KW, Park EK, Hong JT (2017). Parkin deficiency exacerbate ethanol-induced dopaminergic neurodegeneration by P38 pathway dependent inhibition of autophagy and mitochondrial function. Redox Biol 11: 456-468. doi: 10.1016/j.redox.2016.12.008
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, Licitra F, Lopez Ramirez A, Pavel M, Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y, Skidmore J, Rubinsztein DC (2017). Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 93(5): 1015-1034. doi: 10.1016/j.neuron.2017.01.022
- Plaza-Zabala A, Sierra-Torre V, Sierra A (2017). Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int J Mol Sci 18(3): E598. doi: 10.3390/ijms18030598
- Yuan B, Shen H, Lin L, Su T, Zhong L, Yang Z (2017). Autophagy Promotes Microglia Activation Through Beclin-1-Atg5 Pathway in Intracerebral Hemorrhage. Mol Neurobiol 54(1): 115-124. doi: 10.1007/s12035-015-9642-z
- Zhong Z, Sanchez-Lopez E, Karin M (2016). Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin Exp Rheumatol 34(4 Suppl 98): 12-16. PMID: 27586797
- Suh HW, Kim JK, Kim TS, Jo EK (2017). New insights into vitamin D and autophagy in inflammatory bowel diseases. Curr Med Chem 24(9):898-910. doi: 10.2174/0929867323666161202151856
- Miettinen TP, Bjorklund M (2016). The mevalonate pathway as a metabolic requirement for autophagy-implications for growth control, proteostasis, and disease. Mol Cell Oncol 3(3): e1143546. doi: 10.1080/23723556.2016.1143546
- Jia G, Sowers JR (2015). Autophagy: a housekeeper in cardiorenal metabolic health and disease. Biochim Biophys Acta 1852(2): 219-224. doi: 10.1016/j.bbadis.2014.06.025
- Wang F, Jia J, Rodrigues B (2017). Autophagy, Metabolic Disease, and Pathogenesis of Heart Dysfunction. Can J Cardiol 33(7):850-859. doi: 10.1016/j.cjca.2017.01.002
- Zhang S, Lin X, Li G, Shen X, Niu D, Lu G, Fu X, Chen Y, Cui M, Bai Y (2017). Knockout of Eva1a leads to rapid development of heart failure by impairing autophagy. Cell Death Dis 8(2): e2586. doi: 10.1038/cddis.2017.17
- Ren SY, Xu X (2015). Role of autophagy in metabolic syndrome-associated heart disease. Biochim Biophys Acta 1852(2): 225-231. doi: 10.1016/j.bbadis.2014.04.029
- Rubinsztein David C, Mariño G, Kroemer G (2011). Autophagy and Aging. Cell 146(5): 682-695. doi: 10.1016/j.cell.2011.07.030
- Mizushima N, Levine B (2010). Autophagy in mammalian development and differentiation. Nat Cell Biol 12(9): 823-830. doi: 10.1038/ncb0910-823
- Mizushima N, Komatsu M (2011). Autophagy: Renovation of Cells and Tissues. Cell 147(4): 728-741. doi: 10.1016/j.cell.2011.10.026
- Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X (2009). ULK1·ATG13·FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy. J Biol Chem 284(18): 12297-12305. doi: 10.1074/jbc.M900573200
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S-i, Natsume T, Takehana K, Yamada N, Guan J-L, Oshiro N, Mizushima N (2009). Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol Biol Cell 20(7): 1981-1991. doi: 10.1091/mbc.E08-12-1248
- Jung CH, Jun CB, Ro S-H, Kim Y-M, Otto NM, Cao J, Kundu M, Kim D-H (2009). ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol Biol Cell 20(7): 1992-2003. doi: 10.1091/mbc.E08-12-1249
- Laplante M, Sabatini DM (2012). mTOR signaling in growth control and disease. Cell 149(2): 274-293. doi: 10.1016/j.cell.2012.03.017
- Loffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, Campbell DG, Wesselborg S, Alessi DR, Stork B (2011). Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 7(7): 696-706. doi: 10.4161/auto.7.7.15451
- Kim J, Kundu M, Viollet B, Guan K-L (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2): 132-141. doi: 10.1038/ncb2152
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441(7095): 885-889. doi: 10.1038/nature04724
- Dooley Hannah C, Razi M, Polson Hannah E, Girardin Stephen E, Wilson Michael I, Tooze Sharon A (2014). WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12–5-16L1. Mol Cell 55(2): 238-252. doi: 10.1016/j.molcel.2014.05.021
- Furuya N, Yu J, Byfield M, Pattingre S, Levine B (2005). The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 1(1): 46-52. doi: 10.4161/auto.1.1.1542
- Russell RC, Tian Y, Yuan H, Park HW, Chang Y-Y, Kim J, Kim H, Neufeld TP, Dillin A, Guan K-L (2013). ULK1 induces autophagy by phosphorylating Beclin-1 and activating Vps34 lipid kinase. Nat Cell Biol 15(7): 741-750. doi: 10.1038/ncb2757
- Dolzhenko E, van Vugt J, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, Ajay SS, Rajan V, Lajoie BR, Johnson NH, Kingsbury Z, Humphray SJ, Schellevis RD, Brands WJ, Baker M, Rademakers R, Kooyman M, Tazelaar GHP, van Es MA, McLaughlin R, Sproviero W, Shatunov A, Jones A, Al Khleifat A, Pittman A, Morgan S, Hardiman O, Al-Chalabi A, Shaw C, Smith B, et al. (2017). Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res 27(11): 1895-1903. doi: 10.1101/gr.225672.117
- Wang S, Xia P, Rehm M, Fan Z (2015). Autophagy and cell reprogramming. Cell Mol Life Sci 72(9): 1699-1713. doi: 10.1007/s00018-014-1829-3
- Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC (2016). Mammalian Autophagy: How Does It Work? Annu Rev Biochem 85(1): 685-713. doi: 10.1146/annurev-biochem-060815-014556
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, Øvervatn A, Bjørkøy G, Johansen T (2007). p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J Biol Chem 282(33): 24131-24145. doi: 10.1074/jbc.m702824200
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, Stenmark H, Johansen T (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171(4): 603. doi: 10.1083/jcb.200507002
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dötsch V, Bumann D, Dikic I (2011). Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333(6039): 228-233. doi: 10.1126/science.1205405
- Thurston TLM, Ryzhakov G, Bloor S, von Muhlinen N, Randow F (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 10(11): 1215-U1103. doi: 10.1038/ni.1800
- Kirkin V, Lamark T, Sou Y-S, Bjørkøy G, Nunn JL, Bruun J-A, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat J-P, Øvervatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T (2009). A Role for NBR1 in Autophagosomal Degradation of Ubiquitinated Substrates. Mol Cell 33(4): 505-516. doi: 10.1016/j.molcel.2009.01.020
- Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, Bartlett BJ, Myers KM, Birkeland HCG, Lamark T, Krainc D, Brech A, Stenmark H, Simonsen A, Yamamoto A (2010). The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 38(2): 265-279. doi: 10.1016/j.molcel.2010.04.007
- Mandell Michael A, Jain A, Arko-Mensah J, Chauhan S, Kimura T, Dinkins C, Silvestri G, Münch J, Kirchhoff F, Simonsen A, Wei Y, Levine B, Johansen T, Deretic V (2014). TRIM Proteins Regulate Autophagy and Can Target Autophagic Substrates by Direct Recognition. Dev Cell 30(4): 394-409. doi: 10.1016/j.devcel.2014.06.013
- Jäger S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen E-L (2004). Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 117(20): 4837. doi: 10.1242/jcs.01370
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P (2000). Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406(6798): 902-906. doi: 10.1038/35022595
- Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z, Zhang G, Chen Y, Feng D, Hu J, Zhang H (2016). The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol Cell 63(5): 781-795. doi: 10.1016/j.molcel.2016.08.021
- Zhao H, Zhao YG, Wang X, Xu L, Miao L, Feng D, Chen Q, Kovacs AL, Fan D, Zhang H (2013). Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol 200(6): 731-741. doi: 10.1083/jcb.201211014
- Takats S, Pircs K, Nagy P, Varga A, Karpati M, Hegedus K, Kramer H, Kovacs AL, Sass M, Juhasz G (2014). Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol Biol Cell 25(8): 1338-1354. doi: 10.1091/mbc.E13-08-0449
- Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, Mizushima N (2014). The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell 25(8): 1327-1337. doi: 10.1091/mbc.E13-08-0447
- McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I (2015). PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57(1): 39-54. doi: 10.1016/j.molcel.2014.11.006
- Itakura E, Kishi-Itakura C, Mizushima N (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151(6): 1256-1269. doi: 10.1016/j.cell.2012.11.001
- Epple UD, Suriapranata I, Eskelinen E-L, Thumm M (2001). Aut5/Cvt17p, a Putative Lipase Essential for Disintegration of Autophagic Bodies inside the Vacuole. J Bacteriol 183(20): 5942-5955. doi: 10.1128/JB.183.20.5942-5955.2001
- Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E (2005). Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 1(2): 84-91. doi: 10.4161/auto.1.2.1697
- Teter SA, Eggerton KP, Scott SV, Kim J, Fischer AM, Klionsky DJ (2001). Degradation of Lipid Vesicles in the Yeast Vacuole Requires Function of Cvt17, a Putative Lipase. J Biol Chem 276(3): 2083-2087. doi: 10.1074/jbc.C000739200
- Lee S, Sato Y, Nixon RA (2011). Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci 31(21): 7817-7830. doi: 10.1523/JNEUROSCI.6412-10.2011
- Guo F, Liu X, Cai H, Le W (2017). Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol 28(1): 3-13. doi: 10.1111/bpa.12545
- Nixon RA (2013). The role of autophagy in neurodegenerative disease. Nat Med 19(8):983-97. doi: 10.1038/nm.3232
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441(7095): 880-884. doi: 10.1038/nature04723
- Nah J, Yuan J, Jung Y-K (2015). Autophagy in Neurodegenerative Diseases: From Mechanism to Therapeutic Approach. Mol Cells 38(5): 381-389. doi: 10.14348/molcells.2015.0034
- Ravikumar B, Duden R, Rubinsztein DC (2002). Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11(9): 1107-1117. doi: 10.1093/hmg/11.9.1107
- Menzies FM, Fleming A, Rubinsztein DC (2015). Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16: 345. doi: 10.1038/nrn3961
- Yoon S-Y, Kim D-H (2016). Alzheimer's disease genes and autophagy. Brain Res 1649: 201-209. doi: 10.1016/j.brainres.2016.03.018
- Hung SY, Huang WP, Liou HC, Fu WM (2009). Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy 5(4): 502-510. doi: 10.4161/auto.5.4.8096
- Wang H, Ma J, Tan Y, Wang Z, Sheng C, Chen S, Ding J (2010). Amyloid-beta1-42 induces reactive oxygen species-mediated autophagic cell death in U87 and SH-SY5Y cells. JAD 21(2): 597-610. doi: 10.3233/jad-2010-091207
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64(2): 113-122. doi: 10.1093/jnen/64.2.113
- Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido Takaomi C (2013). Aβ Secretion and Plaque Formation Depend on Autophagy. Cell Rep 5(1): 61-69. doi: 10.1016/j.celrep.2013.08.042
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA (2008). Autophagy Induction and Autophagosome Clearance in Neurons: Relationship to Autophagic Pathology in Alzheimer's Disease. J Neurosci 28(27): 6926-6937. doi: 10.1523/JNEUROSCI.0800-08.2008
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V (2010). Inhibition of mTOR by Rapamycin Abolishes Cognitive Deficits and Reduces Amyloid-β Levels in a Mouse Model of Alzheimer's Disease. PLoS ONE 5(4): e9979. doi: 10.1371/journal.pone.0009979
- Tian Y, Bustos V, Flajolet M, Greengard P (2011). A small-molecule enhancer of autophagy decreases levels of Aβ and APP-CTF via Atg5-dependent autophagy pathway. FASEB J 25(6): 1934-1942. doi: 10.1096/fj.10-175158
- Vingtdeux V, Chandakkar P, Zhao H, d'Abramo C, Davies P, Marambaud P (2011). Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-β peptide degradation. FASEB J 5(1): 219-231. doi: 10.1096/fj.10-167361
- Lee VMY, Goedert M, Trojanowski JQ (2001). Neurodegenerative Tauopathies. Annu Rev Neurosci 24(1): 1121-1159. doi: 10.1146/annurev.neuro.24.1.1121
- Majid T, Ali YO, Venkitaramani DV, Jang M-K, Lu H-C, Pautler RG (2014). In vivo axonal transport deficits in a mouse model of fronto-temporal dementia. Neuroimage Clin 4: 711-717. doi: 10.1016/j.nicl.2014.02.005
- Butzlaff M, Hannan SB, Karsten P, Lenz S, Ng J, Voβfeldt H, Prüβing K, Pflanz R, Schulz JB, Rasse T, Voigt A (2015). Impaired retrograde transport by the Dynein/Dynactin complex contributes to Tau-induced toxicity. Hum Mol Genet 24(13): 3623-3637. doi: 10.1093/hmg/ddv107
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S (2010). Molecular Interplay between Mammalian Target of Rapamycin (mTOR), Amyloid-β, and Tau. J Biol Chem 285(17): 13107-13120. doi: 10.1074/jbc.m110.100420
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, Rubinsztein DC (2006). Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet 15(3): 433-442. doi: 10.1093/hmg/ddi458
- Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow E-M, Cuervo AM, Mandelkow E (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18(21): 4153-4170. doi: 10.1093/hmg/ddp367
- Majumder S, Richardson A, Strong R, Oddo S (2011). Inducing Autophagy by Rapamycin Before, but Not After, the Formation of Plaques and Tangles Ameliorates Cognitive Deficits. PLoS ONE 6(9): e25416. doi: 10.1371/journal.pone.0025416
- Rodríguez-Navarro JA, Rodríguez L, Casarejos MJ, Solano RM, Gómez A, Perucho J, Cuervo AM, García de Yébenes J, Mena MA (2010). Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis 39(3): 423-438. doi: 10.1016/j.nbd.2010.05.014
- Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winkler DT, Goedert M (2012). Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 135(7): 2169-2177. doi: 10.1093/brain/aws143
- Perez SE, He B, Nadeem M, Wuu J, Ginsberg SD, Ikonomovic MD, Mufson EJ (2015). Hippocampal Endosomal, Lysosomal and Autophagic Dysregulation in Mild Cognitive Impairment: Correlation with Aβ and Tau Pathology. J Neuropathol Exp Neurol 74(4): 345-358. doi: 10.1097/NEN.0000000000000179
- Collin L, Bohrmann B, Göpfert U, Oroszlan-Szovik K, Ozmen L, Grüninger F (2014). Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer‘s disease. Brain 137(10): 2834-2846. doi: 10.1093/brain/awu213
- Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee J-H, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Näslund J, Mathews PM, Cataldo AM, Nixon RA (2005). Macroautophagy—a novel β-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol 171(1): 87-98. doi: 10.1083/jcb.200505082
- Lee J-H, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Sisodia SS, Cuervo AM, Nixon RA (2010). Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 141(7): 1146-1158. doi: 10.1016/j.cell.2010.05.008
- Bustos V, Pulina MV, Bispo A, Lam A, Flajolet M, Gorelick FS, Greengard P (2017). Phosphorylated Presenilin 1 decreases β-amyloid by facilitating autophagosome–lysosome fusion. Proc Natl Acad Sci U S A 114(27): 7148-7153. doi: 10.1073/pnas.1705240114
- Yang D-S, Stavrides P, Saito M, Kumar A, Rodriguez-Navarro JA, Pawlik M, Huo C, Walkley SU, Saito M, Cuervo AM, Nixon RA (2014). Defective macroautophagic turnover of brain lipids in the TgCRND8 Alzheimer mouse model: prevention by correcting lysosomal proteolytic deficits. Brain 137(12): 3300-3318. doi: 10.1093/brain/awu278
- Tammineni P, Ye X, Feng T, Aikal D, Cai Q (2017). Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer's disease neurons. eLife 6: e21776. doi: 10.7554/eLife.21776
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest 118(6): 2190-2199. doi: 10.1172/JCI33585
- Small Scott A, Kent K, Pierce A, Leung C, Kang Min S, Okada H, Honig L, Vonsattel JP, Kim TW (2005). Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol 58(6): 909-919. doi: 10.1002/ana.20667
- Rohn TT, Wirawan E, Brown RJ, Harris JR, Masliah E, Vandenabeele P (2011). Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer's disease brain. Neurobiol Dis 43(1): 68-78. doi: 10.1016/j.nbd.2010.11.003
- Pajares M, Jiménez-Moreno N, García-Yagüe ÁJ, Escoll M, de Ceballos ML, Van Leuven F, Rábano A, Yamamoto M, Rojo AI, Cuadrado A (2016). Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 12(10): 1902-1916. doi: 10.1080/15548627.2016.1208889
- Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GVW (2014). Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun 5: 3496-3496. doi: 10.1038/ncomms4496
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere M, Singh Pahwa J, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan A, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown K, Passmore P, Craig D, McGuinness B, Todd S, Holmes C, et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease, and shows evidence for additional susceptibility genes. Nat Genet 41(10): 1088-1093. doi: 10.1038/ng.440
- Jun G, Naj AC, Beecham GW, Wang L-S, Buros J, Gallins PJ, Buxbaum JD, Ertekin-Taner N, Fallin MD, Friedland R, Inzelberg R, Kramer P, Rogaeva E, St George-Hyslop P, Adgc, Cantwell LB, Dombroski BA, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Lunetta KL, Martin ER, Montine TJ, Goate AM, Blacker D, Tsuang DW, Beekly D, Cupples LA, Hakonarson H, et al. (2010). Meta-Analysis confirms CR1, CLU, and PICALM as Alzheimer's disease risk loci and reveals interactions with APOE genotypes. Arch Neurol 67(12): 1473-1484. doi: 10.1001/archneurol.2010.201
- Ando K, Brion J-P, Stygelbout V, Suain V, Authelet M, Dedecker R, Chanut A, Lacor P, Lavaur J, Sazdovitch V, Rogaeva E, Potier M-C, Duyckaerts C (2013). Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer's brains. Acta Neuropathol 125(6): 861-878. doi: 10.1007/s00401-013-1111-z
- Ando K, Tomimura K, Sazdovitch V, Suain V, Yilmaz Z, Authelet M, Ndjim M, Vergara C, Belkouch M, Potier M-C, Duyckaerts C, Brion J-P (2016). Level of PICALM, a key component of clathrin-mediated endocytosis, is correlated with levels of phosphotau and autophagy-related proteins and is associated with tau inclusions in AD, PSP and Pick disease. Neurobiol Dis 94: 32-43. doi: 10.1016/j.nbd.2016.05.017
- Moreau K, Fleming A, Imarisio S, Lopez Ramirez A, Mercer JL, Jimenez-Sanchez M, Bento CF, Puri C, Zavodszky E, Siddiqi F, Lavau CP, Betton M, O'Kane CJ, Wechsler DS, Rubinsztein DC (2014). PICALM modulates autophagy activity and tau accumulation. Nat Commun 5: 4998. doi: 10.1038/ncomms5998
- Tian Y, Chang JC, Fan EY, Flajolet M, Greengard P (2013). Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer's APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci U S A 110(42): 17071-17076. doi: 10.1073/pnas.1315110110
- Yang C, Cai C-Z, Song J-X, Tan J-Q, Durairajan SSK, Iyaswamy A, Wu M-Y, Chen L-L, Yue Z, Li M, Lu J-H (2017). NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 13(12): 2028-2040. doi: 10.1080/15548627.2017.1379633
- Zhang L, Fang Y, Cheng X, Lian Y, Xu H, Zeng Z, Zhu H (2017). TRPML1 Participates in the Progression of Alzheimer's Disease by Regulating the PPARγ/AMPK/Mtor Signalling Pathway. Cell Physiol Biochem 43(6): 2446-2456. doi: 10.1159/000484449
- Ulland TK, Song WM, Huang SC-C, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M (2017). TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease. Cell 170(4): 649-663.e613. doi: 10.1016/j.cell.2017.07.023
- Chen K, Yuan R, Geng S, Zhang Y, Ran T, Kowalski E, Liu J, Li L (2017). Toll-interacting protein deficiency promotes neurodegeneration via impeding autophagy completion in high-fat diet-fed ApoE−/− mouse model. Brain Behav Immun 59: 200-210. doi: 10.1016/j.bbi.2016.10.002
- Wang B-J, Her GM, Hu M-K, Chen Y-W, Tung Y-T, Wu P-Y, Hsu W-M, Lee H, Jin L-W, Hwang S-PL, Chen RPY, Huang C-J, Liao Y-F (2017). ErbB2 regulates autophagic flux to modulate the proteostasis of APP-CTFs in Alzheimer's disease. Proc Natl Acad Sci U S A 114(15): E3129-E3138. doi: 10.1073/pnas.1618804114
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PloS one 5(4): e9979-e9979. doi: 10.1371/journal.pone.0009979
- Zhu Z, Yan J, Jiang W, Yao XG, Chen J, Chen L, Li C, Hu L, Jiang H, Shen X (2013). Arctigenin Effectively Ameliorates Memory Impairment in Alzheimer's Disease Model Mice Targeting Both-Amyloid Production and Clearance. J Neurosci 33(32): 13138-13149. doi: 10.1523/jneurosci.4790-12.2013
- Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, Marambaud P (2010). AMP-activated Protein Kinase Signaling Activation by Resveratrol Modulates Amyloid-β Peptide Metabolism. J Biol Chem 285(12): 9100-9113. doi: 10.1074/jbc.m109.060061
- Xue X, Wang L-R, Sato Y, Jiang Y, Berg M, Yang D-S, Nixon RA, Liang X-J (2014). Single-Walled Carbon Nanotubes Alleviate Autophagic/Lysosomal Defects in Primary Glia from a Mouse Model of Alzheimer's Disease. Nano Lett 14(9): 5110-5117. doi: 10.1021/nl501839q
- Chu C, Zhang X, Ma W, Li L, Wang W, Shang L, Fu P (2013). Induction of autophagy by a novel small molecule improves abeta pathology and ameliorates cognitive deficits. PLoS One 8(6): e65367. doi: 10.1371/journal.pone.0065367
- Steele JW, Lachenmayer ML, Ju S, Stock A, Liken J, Kim SH, Delgado LM, Alfaro IE, Bernales S, Verdile G, Bharadwaj P, Gupta V, Barr R, Friss A, Dolios G, Wang R, Ringe D, Fraser P, Westaway D, St George-Hyslop PH, Szabo P, Relkin NR, Buxbaum JD, Glabe CG, Protter AA, Martins RN, Ehrlich ME, Petsko GA, Yue Z, Gandy S (2013). Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer's mouse model. Mol Psychiatry 18(8): 889-897. doi: 10.1038/mp.2012.106
- Parr C, Carzaniga R, Gentleman SM, Van Leuven F, Walter J, Sastre M (2012). Glycogen Synthase Kinase 3 Inhibition Promotes Lysosomal Biogenesis and Autophagic Degradation of the Amyloid-Precursor Protein. Mol Cell Biol 32(21): 4410-4418. doi: 10.1128/mcb.00930-12
- Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H (2013). Inhibition of Glycogen Synthase Kinase-3 Ameliorates β-Amyloid Pathology and Restores Lysosomal Acidification and Mammalian Target of Rapamycin Activity in the Alzheimer Disease Mouse Model. J Biol Chem 288(2): 1295-1306. doi: 10.1074/jbc.m112.409250
- Jiang T, Yu J-T, Zhu X-C, Tan M-S, Wang H-F, Cao L, Zhang Q-Q, Shi J-Q, Gao L, Qin H, Zhang Y-D, Tan L (2014). Temsirolimus promotes autophagic clearance of amyloid-β and provides protective effects in cellular and animal models of Alzheimer's disease. Pharmacol Res 81: 54-63. doi: 10.1016/j.phrs.2014.02.008
- Lonskaya I, Hebron ML, Desforges NM, Schachter JB, Moussa CEH (2014). Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J Mol Med 92(4): 373-386. doi: 10.1007/s00109-013-1112-3
- Guo X, Lv J, Lu J, Fan L, Huang X, Hu L, Wang J, Shen X (2018). Protopanaxadiol derivative DDPU improves behavior and cognitive deficit in AD mice involving regulation of both ER stress and autophagy. Neuropharmacology 130: 77-91. doi: 10.1016/j.neuropharm.2017.11.033
- Huang M, Jiang X, Liang Y, Liu Q, Chen S, Guo Y (2017). Berberine improves cognitive impairment by promoting autophagic clearance and inhibiting production of beta-amyloid in APP/tau/PS1 mouse model of Alzheimer's disease. Exp Gerontol 91: 25-33. doi: 10.1016/j.exger.2017.02.004
- Li LS, Lu YL, Nie J, Xu YY, Zhang W, Yang WJ, Gong QH, Lu YF, Lu Y, Shi JS (2017). Dendrobium nobile Lindl alkaloid, a novel autophagy inducer, protects against axonal degeneration induced by Abeta25-35 in hippocampus neurons in vitro. CNS Neurosci Ther 23(4): 329-340. doi: 10.1111/cns.12678
- Dauer W, Przedborski S (2003). Parkinson's Disease. Neuron 39(6): 889-909. doi: 10.1016/s0896-6273(03)00568-3
- Moloudizargari M, Asghari MH, Ghobadi E, Fallah M, Rasouli S, Abdollahi M (2017). Autophagy, its mechanisms and regulation: Implications in neurodegenerative diseases. Ageing Res Rev 40: 64-74. doi: 10.1016/j.arr.2017.09.005
- Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, Vila M (2010). Pathogenic Lysosomal Depletion in Parkinson's Disease. J Neurosci 30(37): 12535. doi: 10.1523/jneurosci.1920-10.2010
- Dauer W, Przedborski S (2003). Parkinson's Disease: Mechanisms and Models. Neuron 39(6): 889-909. doi: 10.1016/S0896-6273(03)00568-3
- Kalia LV, Kalia SK, McLean PJ, Lozano AM, Lang AE (2013). α-Synuclein oligomers and clinical implications for Parkinson disease. Ann Neurol 73(2): 155-169. doi: 10.1002/ana.23746
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004). Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 305(5688): 1292. doi: 10.1126/science.1101738
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC (2003). Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278(27): 25009-25013. doi: 10.1074/jbc.M300227200
- Winslow AR, Chen C-W, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC (2010). α-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol 190(6): 1023-1037. doi: 10.1083/jcb.201003122
- Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L (2009). Abberant α-Synuclein Confers Toxicity to Neurons in Part through Inhibition of Chaperone-Mediated Autophagy. PLoS ONE 4(5): e5515. doi: 10.1371/journal.pone.0005515
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011). TFEB links autophagy to lysosomal biogenesis. Science 332(6036): 1429-1433. doi: 10.1126/science.1204592
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Björklund A (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci U S A 110(19): E1817-E1826. doi: 10.1073/pnas.1305623110
- Kilpatrick K, Zeng Y, Hancock T, Segatori L (2015). Genetic and Chemical Activation of TFEB Mediates Clearance of Aggregated α-Synuclein. PLoS ONE 10(3): e0120819. doi: 10.1371/journal.pone.0120819
- Zavodszky E, Seaman MNJ, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, Rubinsztein DC (2014). Mutation in VPS35 associated with Parkinson's disease impairs WASH complex association and inhibits autophagy. Nat Commun 5: 3828-3828. doi: 10.1038/ncomms4828
- Skibinski G, Nakamura K, Cookson MR, Finkbeiner S (2014). Mutant LRRK2 Toxicity in Neurons Depends on LRRK2 Levels and Synuclein But Not Kinase Activity or Inclusion Bodies. J Neurosci 34(2): 418-433. doi: 10.1523/JNEUROSCI.2712-13.2014
- Plowey ED, Cherra SJ, Liu Y-J, Chu CT (2008). Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 105(3): 1048-1056. doi: 10.1111/j.1471-4159.2008.05217.x
- Saha S, Ash PEA, Gowda V, Liu L, Shirihai O, Wolozin B (2015). Mutations in LRRK2 potentiate age-related impairment of autophagic flux. Mol Neurodegener 10: 26. doi: 10.1186/s13024-015-0022-y
- Hartlova A, Herbst S, Peltier J, Rodgers A, Bilkei-Gorzo O, Fearns A, Dill BD, Lee H, Flynn R, Cowley SA, Davies P, Lewis PA, Ganley IG, Martinez J, Alessi DR, Reith AD, Trost M, Gutierrez MG, Hwang CJ, Kim YE, Son DJ, Park MH, Choi D-Y, Park P-H, Hellström M, Han S-B, Oh K-W, Park EK, Hong JT (2018). LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J 37(12): e98694. doi: 10.15252/embj.201798694
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392(6676): 605-608. doi: 10.1038/33416
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004). Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304(5674): 1158-60. doi: 10.1126/science.1096284
- Kazlauskaite A, Muqit MMK (2015). PINK1 and Parkin – mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson's disease. Febs J 282(2): 215-223. doi: 10.1111/febs.13127
- Kitada T, Pisani A, Karouani M, Haburcak M, Martella G, Tscherter A, Platania P, Wu B, Pothos Emmanuel N, Shen J (2009). Impaired dopamine release and synaptic plasticity in the striatum of Parkin−/− mice. J Neurochem 110(2): 613-621. doi: 10.1111/j.1471-4159.2009.06152.x
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J (2004). Mitochondrial Dysfunction and Oxidative Damage in parkin-deficient Mice. J Biol Chem 279(18): 18614-18622. doi: 10.1074/jbc.M401135200
- Gautier CA, Kitada T, Shen J (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105(32): 11364-11369. doi: 10.1073/pnas.0802076105
- Narendra D, Tanaka A, Suen D-F, Youle RJ (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183(5): 795-803. doi: 10.1083/jcb.200809125
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RLA, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107(1): 378-383. doi: 10.1073/pnas.0911187107
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou Y-s, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189(2): 211-221. doi: 10.1083/jcb.200910140
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe J-F, Saeki Y, Tanaka K, Matsuda N (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162. doi: 10.1038/nature13392
- Narendra DP, Jin SM, Tanaka A, Suen D-F, Gautier CA, Shen J, Cookson MR, Youle RJ (2010). PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol 8(1): e1000298. doi: 10.1371/journal.pbio.1000298
- Shim JH, Yoon SH, Kim K-H, Han JY, Ha J-Y, Hyun DH, Paek SH, Kang UJ, Zhuang X, Son JH (2011). The antioxidant Trolox helps recovery from the familial Parkinson's disease-specific mitochondrial deficits caused by PINK1- and DJ-1-deficiency in dopaminergic neuronal cells. Mitochondrion 11(5): 707-715. doi: 10.1016/j.mito.2011.05.013
- De Miranda BR, Rocha EM, Bai Q, El Ayadi A, Hinkle D, Burton EA, Timothy Greenamyre J (2018). Astrocyte-specific DJ-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of Parkinson's disease. Neurobiol Dis 115: 101-114. doi: 10.1016/j.nbd.2018.04.008
- Wang X, Zhai H, Wang F (2018). 6-OHDA Induces Oxidation of F-box Protein Fbw7β by Chaperone-Mediated Autophagy in Parkinson's Model. Mol Neurobiol 55(6): 4825-4833. doi: 10.1007/s12035-017-0686-0
- Abeliovich A, Gitler AD (2016). Defects in trafficking bridge Parkinson's disease pathology and genetics. Nature 539(7628): 207-216. doi: 10.1038/nature20414
- Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, Cooper A, Garner B, Halliday GM (2014). Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson's disease. Brain 137(3): 834-848. doi: 10.1093/brain/awt367
- Djarmati A, Hagenah J, Reetz K, Winkler S, Behrens Maria I, Pawlack H, Lohmann K, Ramirez A, Tadić V, Brüggemann N, Berg D, Siebner Hartwig R, Lang Anthony E, Pramstaller Peter P, Binkofski F, Kostić Vladimir S, Volkmann J, Gasser T, Klein C (2009). ATP13A2 variants in early-onset Parkinson's disease patients and controls. Mov Disord 24(14): 2104-2111. doi: 10.1002/mds.22728
- Ramirez A, Heimbach A, Gruendemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38(10): 1184-1191. doi: 10.1038/ng1884
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron M-H, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E (2012). Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109(24): 9611-9616. doi: 10.1073/pnas.1112368109
- Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D (2012). Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation and neurotoxicity. J Neurosci 32(12): 4240-4246. doi: 10.1523/JNEUROSCI.5575-11.2012
- Bento CF, Ashkenazi A, Jimenez-Sanchez M, Rubinsztein DC (2016). The Parkinson's disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat Commun 7: 11803. doi: 10.1038/ncomms11803
- Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun Sidi M, Pujol C, Ciura S, Erpapazoglou Z, Usenko T, Maurage C-A, Sahbatou M, Liebau S, Ding J, Bilgic B, Emre M, Erginel-Unaltuna N, Guven G, Tison F, Tranchant C, Vidailhet M, Corvol J-C, Krack P, Leutenegger A-L, Nalls Michael A, Hernandez Dena G, Heutink P, Gibbs JR, et al. (2016). Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am J Hum Genet 98(3): 500-513. doi: 10.1016/j.ajhg.2016.01.014
- Gleich K, Desmond MJ, Lee D, Berkovic SF, Dibbens LM, Katerelos M, Bayly MA, Fraser SA, Martinello P, Vears DF, Mount P, Power DA (2013). Abnormal Processing of Autophagosomes in Transformed B Lymphocytes from SCARB2-Deficient Subjects. BioRes Open Access 2(1): 40-46. doi: 10.1089/biores.2012.0265
- Gonzalez A, Valeiras M, Sidransky E, Tayebi N (2014). Lysosomal Integral Membrane Protein-2: A New Player in Lysosome-Related Pathology. Mol Genet Metab 111(2): 84-91. doi: 10.1016/j.ymgme.2013.12.005
- Janda E, Isidoro C, Carresi C, Mollace V (2012). Defective Autophagy in Parkinson's Disease: Role of Oxidative Stress. Mol Neurobiol 46(3): 639-661. doi: 10.1007/s12035-012-8318-1
- Choi K-C, Kim S-H, Ha J-Y, Kim S-T, Son Jin H (2009). A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J Neurochem 112(2): 366-376. doi: 10.1111/j.1471-4159.2009.06463.x
- Jinn S, Drolet RE, Cramer PE, Wong AH-K, Toolan DM, Gretzula CA, Voleti B, Vassileva G, Disa J, Tadin-Strapps M, Stone DJ (2017). TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc Natl Acad Sci U S A 114(9): 2389-2394. doi: 10.1073/pnas.1616332114
- Wu Y, Li X, Zhu JX, Xie W, Le W, Fan Z, Jankovic J, Pan T (2011). Resveratrol-Activated AMPK/SIRT1/Autophagy in Cellular Models of Parkinson's Disease. Neurosignals 19(3): 163-174. doi: 10.1159/000328516
- Jiang T-F, Zhang Y-J, Zhou H-Y, Wang H-M, Tian L-P, Liu J, Ding J-Q, Chen S-D (2013). Curcumin Ameliorates the Neurodegenerative Pathology in A53T α-synuclein Cell Model of Parkinson's Disease Through the Downregulation of mTOR/p70S6K Signaling and the Recovery of Macroautophagy. J Neuroimmune Pharmacol 8(1): 356-369. doi: 10.1007/s11481-012-9431-7
- Lan D-M, Liu F-T, Zhao J, Chen Y, Wu J-J, Ding Z-T, Yue Z-Y, Ren H-M, Jiang Y-P, Wang J (2012). Effect of Trehalose on PC12 Cells Overexpressing Wild-Type or A53T Mutant α-synuclein. Neurochem Res 37(9): 2025-2032. doi: 10.1007/s11064-012-0823-0
- Hebron ML, Lonskaya I, Moussa CEH (2013). Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson's disease models. Hum Mol Genet 22(16): 3315-3328. doi: 10.1093/hmg/ddt192
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E (2009). Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci 29(43): 13578-13588. doi: 10.1523/JNEUROSCI.4390-09.2009
- Liu J, Chen M, Wang X, Wang Y, Duan C, Gao G, Lu L, Wu X, Wang X, Yang H (2016). Piperine induces autophagy by enhancing protein phosphotase 2A activity in a rotenone-induced Parkinson's disease model. Oncotarget 7(38): 60823-60843. doi: 10.18632/oncotarget.11661
- Hou YS, Guan JJ, Xu HD, Wu F, Sheng R, Qin ZH (2015). Sestrin2 Protects Dopaminergic Cells against Rotenone Toxicity through AMPK-Dependent Autophagy Activation. Mol Cell Biol 35(16): 2740-2751. doi: 10.1128/MCB.00285-15
- Yang G, Li J, Cai Y, Yang Z, Li R, Fu W (2018). Glycyrrhizic Acid Alleviates 6-Hydroxydopamine and Corticosterone-Induced Neurotoxicity in SH-SY5Y Cells Through Modulating Autophagy. Neurochem Res 43(10): 1914-1926. doi: 10.1007/s11064-018-2609-5
- Jang W, Ju Kim H, Li H, Jo K, Kyu Lee M, Hong Song S, Ok Yang H (2014). 1,25-Dyhydroxyvitamin D-3 attenuates rotenone-induced neurotoxicity in SH-SY5Y cells through induction of autophagy. Biochem Biophys Res Commun 451(1):142-7. doi: 10.1016/j.bbrc.2014.07.081
- Imarisio S, Carmichael J, Korolchuk V, Chen C-W, Saiki S, Rose C, Krishna G, Davies Janet E, Ttofi E, Underwood Benjamin R, Rubinsztein David C (2008). Huntington's disease: from pathology and genetics to potential therapies. Biochem J 412(2): 191. doi: 10.1042/BJ20071619
- Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC (2017). Huntington's Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb Perspect Med 7(7): a024240. doi: 10.1101/cshperspect.a024240
- Dayalu P, Albin RL (2015). Huntington Disease: Pathogenesis and Treatment. Neurol Clin 33(1): 101-114. doi: 10.1016/j.ncl.2014.09.003
- Tellez-Nagel I, Johnson AB, Terry RD (1974). Studies on brain biopsies of patients with Huntington's chorea. J Neuropathol Exp Neurol 33(2): 308-332. doi: 10.1097/00005072-197404000-00008
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36(6): 585-595. doi: 10.1038/ng1362
- Martin DDO, Ladha S, Ehrnhoefer DE, Hayden MR (2015). Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci 38(1): 26-35. doi: 10.1016/j.tins.2014.09.003
- Metzger S, Saukko M, Van Che H, Tong L, Puder Y, Riess O, Nguyen HP (2010). Age at onset in Huntington's disease is modified by the autophagy pathway: implication of the V471A polymorphism in Atg7. Hum Genet 128(4): 453-459. doi: 10.1007/s00439-010-0873-9
- Martinez-Vicente M, Talloczy Z, Wong E, Tang GM, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci 13(5): 567-U574. doi: 10.1038/nn.2528
- Zheng S, Clabough EBD, Sarkar S, Futter M, Rubinsztein DC, Zeitlin SO (2010). Deletion of the Huntingtin Polyglutamine Stretch Enhances Neuronal Autophagy and Longevity in Mice. PLoS Genet 6(2): e1000838. doi: 10.1371/journal.pgen.1000838
- Atwal RS, Truant R (2008). A stress sensitive ER membrane-association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy. Autophagy 4(1): 91-93. doi: 10.4161/auto.5201
- Zheng S, Clabough EB, Sarkar S, Futter M, Rubinsztein DC, Zeitlin SO (2010). Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet 6(2): e1000838. doi: 10.1371/journal.pgen.1000838
- Rui Y-N, Xu Z, Patel B, Chen Z, Chen D, Tito A, David G, Sun Y, Stimming EF, Bellen HJ, Cuervo AM, Zhang S (2015). Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol 17(3): 262-275. doi: 10.1038/ncb3101
- Walter C, Clemens LE, Muller AJ, Fallier-Becker P, Proikas-Cezanne T, Riess O, Metzger S, Nguyen HP (2016). Activation of AMPK-induced autophagy ameliorates Huntington disease pathology in vitro. Neuropharmacology 108: 24-38. doi: 10.1016/j.neuropharm.2016.04.041
- Martinet W, Agostinis P, Vanhoecke B, Dewaele M, De Meyer GR (2009). Autophagy in disease: a double-edged sword with therapeutic potential. Clin Sci 116(9): 697-712. doi: 10.1042/cs20080508
- Squitieri F, Gellera C, Cannella M, Mariotti C, Cislaghi G, Rubinsztein DC, Almqvist EW, Turner D, Bachoud-Levi AC, Simpson SA, Delatycki M, Maglione V, Hayden MR, Donato SD (2003). Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain 126(Pt 4): 946-955. doi: 10.1093/brain/awg077
- Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, Squitieri F, Hardenberg MC, Imarisio S, Menzies FM, Rubinsztein DC (2017). Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 545(7652): 108-111. doi: 10.1038/nature22078
- Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J (2006). Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 281(20): 14474-14485. doi: 10.1074/jbc.M600364200
- Sarkar S, Rubinsztein DC (2008). Huntington's disease: degradation of mutant huntingtin by autophagy. FEBS J 275(17): 4263-4270. doi: 10.1111/j.1742-4658.2008.06562.x
- Wong YC, Holzbaur EL (2014). The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci 34(4): 1293-1305. doi: 10.1523/jneurosci.1870-13.2014
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci 13(5): 567-576. doi: 10.1038/nn.2528
- Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR (2015). Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci 38(1): 26-35. doi: 10.1016/j.tins.2014.09.003
- Lee H, Noh JY, Oh Y, Kim Y, Chang JW, Chung CW, Lee ST, Kim M, Ryu H, Jung YK (2012). IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum Mol Genet 21(1): 101-114. doi: 10.1093/hmg/ddr445
- Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, Squitieri F, Hardenberg MC, Imarisio S, Menzies FM, Rubinsztein DC (2017). Polyglutamine tracts regulate autophagy. Autophagy 13(9): 1613-1614. doi: 10.1080/15548627.2017.1336278
- Mealer RG, Murray AJ, Shahani N, Subramaniam S, Snyder SH (2014). Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J Biol Chem 289(6): 3547-3554. doi: 10.1074/jbc.M113.536912
- Alves S, Cormier-Dequaire F, Marinello M, Marais T, Muriel MP, Beaumatin F, Charbonnier-Beaupel F, Tahiri K, Seilhean D, El HK, Ruberg M, Stevanin G, Barkats M, den Dunnen W, Priault M, Brice A, Durr A, Corvol JC, Sittler A (2014). The autophagy/lysosome pathway is impaired in SCA7 patients and SCA7 knock-in mice. Acta Neuropathol 128(5): 705-722. doi: 10.1007/s00401-014-1289-8
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N (2011). Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 44(2): 279-289. doi: 10.1016/j.molcel.2011.07.039
- Al-Ramahi I, Giridharan S, Chen YC, Patnaik S, Safren N, Hasegawa J, de Haro M, Wagner GA, Titus SA, Jeong H, Clarke J, Krainc D, Zheng W, Irvine RF, Barmada S, Ferrer M, Southall N, Weisman LS, Botas J, Marugan JJ (2017). Inhibition of PIP4Kgamma ameliorates the pathological effects of mutant huntingtin protein. Elife 6: e29123. doi: 10.7554/eLife.29123
- Rai A, Singh PK, Singh V, Kumar V, Mishra R, Thakur AK, Mahadevan A, Shankar SK, Jana NR, Ganesh S (2018). Glycogen synthase protects neurons from cytotoxicity of mutant huntingtin by enhancing the autophagy flux. Cell Death Dis 9(2). doi: 10.1038/s41419-017-0190-5
- Croce KR, Yamamoto A (2018). A role for autophagy in Huntington's disease. Neurobiol Dis 122:16-22. doi: 10.1016/j.nbd.2018.08.010
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC (2007). Trehalose, a Novel mTOR-independent Autophagy Enhancer, Accelerates the Clearance of Mutant Huntingtin and α-Synuclein. J Biol Chem 282(8): 5641-5652. doi: 10.1074/jbc.M609532200
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC (2005). Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 170(7): 1101-1111. doi: 10.1083/jcb.200504035
- Jeong H, Then F, Melia TJ, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D (2009). Acetylation Targets Mutant Huntingtin to Autophagosomes for Degradation. Cell 137(1): 60-72. doi: 10.1016/j.cell.2009.03.018
- Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, Qin ZH (2012). The role of chaperone-mediated autophagy in huntingtin degradation. PLoS One 7(10): e46834. doi: 10.1371/journal.pone.0046834
- Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadiq O, Brown SD, Rubinsztein DC (2010). Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington's disease. Hum Mol Genet 19(11): 2144-2153. doi: 10.1093/hmg/ddq093
- Jia H, Kast RJ, Steffan JS, Thomas EA (2012). Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington's disease mice: implications for the ubiquitin–proteasomal and autophagy systems. Hum Mol Genet 21(24): 5280-5293. doi: 10.1093/hmg/dds379
- Abd-Elrahman KS, Hamilton A, Hutchinson SR, Liu F, Russell RC, Ferguson SSG (2017). mGluR5 antagonism increases autophagy and prevents disease progression in the zQ175 mouse model of Huntington's disease. Sci Signal 10(510): eaan6387. doi: 10.1126/scisignal.aan6387
- Chang C-C, Lin T-C, Ho H-L, Kuo C-Y, Li H-H, Korolenko TA, Chen W-J, Lai T-J, Ho Y-J, Lin C-L (2018). GLP-1 Analogue Liraglutide Attenuates Mutant Huntingtin-Induced Neurotoxicity by Restoration of Neuronal Insulin Signaling. Int J Mol Sci 19(9): 2505. doi: 10.3390/ijms19092505
- Wong VKW, Wu AG, Wang JR, Liu L, Law BY-K (2015). Neferine attenuates the protein level and toxicity of mutant huntingtin in PC-12 cells via induction of autophagy. Molecules 20(3): 3496-3514. doi: 10.3390/molecules20033496
- Hardiman O, van den Berg LH, Kiernan MC (2011). Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 7(11): 639-649. doi: 10.1038/nrneurol.2011.153
- Andersen PM, Al-Chalabi A (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol 7(11): 603. doi: 10.1038/nrneurol.2011.150
- Blokhuis AM, Groen EJN, Koppers M, van den Berg LH, Pasterkamp RJ (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol 125(6): 777-794. doi: 10.1007/s00401-013-1125-6
- Boillee S, Vande Velde C, Cleveland DW (2006). ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 52(1): 39-59. doi: 10.1016/j.neuron.2006.09.018
- Sasaki S (2011). Autophagy in Spinal Cord Motor Neurons in Sporadic Amyotrophic Lateral Sclerosis. J Neuropathol Exp Neurol 70(5): 349-359. doi: 10.1097/NEN.0b013e3182160690
- Li L, Zhang X, Le W (2008). Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 4(3): 290–293. doi: 10.4161/auto.5524
- Morimoto N, Nagai M, Ohta Y, Miyazaki K, Kurata T, Morimoto M, Murakami T, Takehisa Y, Ikeda Y, Kamiya T, Abe K (2007). Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res 1167: 112-117. doi: 10.1016/j.brainres.2007.06.045
- Sheng YW, Chattopadhyay M, Whitelegge J, Valentine JS (2012). SOD1 Aggregation and ALS: Role of Metallation States and Disulfide Status. Curr Top Med Chem 12(22): 2560-2572. doi: 10.2174/15680266112129990079
- An T, Shi P, Duan W, Zhang S, Yuan P, Li Z, Wu D, Xu Z, Li C, Guo Y (2014). Oxidative Stress and Autophagic Alteration in Brainstem of SOD1-G93A Mouse Model of ALS. Mol Neurobiol 49(3): 1435-1448. doi: 10.1007/s12035-013-8623-3
- Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, Piersaint JA, Tapia JC, Rich MM, Maniatis T (2017). Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc Natl Acad Sci U S A 114(39): E8294-E8303. doi: 10.1073/pnas.1704294114
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 23(19): 2294-2306. doi: 10.1101/gad.1830709
- Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E, Galbiati M, Fontana E, Marino M, Carra S, Bendotti C, De Biasi S, Poletti A (2010). The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet 19(17): 3440-3456. doi: 10.1093/hmg/ddq257
- Gal J, Ström A-L, Kwinter DM, Kilty R, Zhang J, Shi P, Fu W, Wooten MW, Zhu H (2009). Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem 111(4): 1062-1073. doi: 10.1111/j.1471-4159.2009.06388.x
- Goode A, Butler K, Long J, Cavey J, Scott D, Shaw B, Sollenberger J, Gell C, Johansen T, Oldham NJ, Searle MS, Layfield R (2016). Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 12(7): 1094–1104. doi: 10.1080/15548627.2016.1170257
- Brady OA, Meng P, Zheng Y, Mao Y, Hu F (2011). Regulation of TDP-43 aggregation by phosphorylation andp62/SQSTM1. J Neurochem 116(2): 248-259. doi: 10.1111/j.1471-4159.2010.07098.x
- Brady OA, Meng P, Zheng YQ, Mao YX, Hu FH (2011). Regulation of TDP-43 aggregation by phosphorylation andp62/SQSTM1. J Neurochem 116(2): 248-259. doi: 10.1111/j.1471-4159.2010.07098.x
- Li F, Xie X, Wang Y, Liu J, Cheng X, Guo Y, Gong Y, Hu S, Pan L (2016). Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat Commun 7: 12708. doi: 10.1038/ncomms12708
- Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, Bruun J-A, Hansen TE, Johansen T, Deretic V (2012). TBK-1 Promotes Autophagy-Mediated Antimicrobial Defense by Controlling Autophagosome Maturation. Immunity 37(2): 223–234. doi: 10.1016/j.immuni.2012.04.015
- Moore AS, Erika LFH (2016). Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A 113(24): E3349. doi: 10.1073/pnas.1523810113
- Sundaramoorthy V, Walker AK, Tan V, Fifita JA, Mccann EP, Williams KL, Blair IP, Guillemin GJ, Farg MA, Atkin JD (2015). Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum Mol Genet 24(13): 3830-3846. doi: 10.1093/hmg/ddv126
- Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick-Jones J, Buss F (2012). Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol 14(10): 1024–1035. doi: 10.1038/ncb2589
- Wong YC, Erika LFH (2014). Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A 111(42): E4439. doi: 10.1073/pnas.1405752111
- Wong YC, Holzbaur ELF (2015). Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 11(2): 422–424. doi: 10.1080/15548627.2015.1009792
- Jantrapirom S, Lo PL, Yoshida H, Yamaguchi M (2018). Depletion of Ubiquilin induces an augmentation in soluble ubiquitinated Drosophila TDP-43 to drive neurotoxicity in the fly. Biochim Biophys Acta Mol Basis Dis 1864(9 Pt B): 3038-3049. doi: 10.1016/j.bbadis.2018.06.017
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477(7363): 211-215. doi: 10.1038/nature10353
- Le NT, Chang L, Kovlyagina I, Georgiou P, Safren N, Braunstein KE, Kvarta MD, Van Dyke AM, LeGates TA, Philips T, Morrison BM, Thompson SM, Puche AC, Gould TD, Rothstein JD, Wong PC, Monteiro MJ (2016). Motor neuron disease, TDP-43 pathology, and memory deficits in mice expressing ALS-FTD-linked UBQLN2 mutations. Proc Natl Acad Sci U S A 113(47): E7580-E7589. doi: 10.1073/pnas.1608432113
- Chang L, Monteiro MJ (2015). Defective Proteasome Delivery of Polyubiquitinated Proteins by Ubiquilin-2 Proteins Containing ALS Mutations. PLoS One 10(6): e0130162. doi: 10.1371/journal.pone.0130162
- Osaka M, Ito D, Suzuki N (2016). Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem Biophys Res Commun472(2): 324-331. doi: 10.1016/j.bbrc.2016.02.107
- Todd TW, Petrucelli L (2016). Insights into the pathogenic mechanisms of Chromosome 9 open reading frame 72 (C9orf72) repeat expansions. J Neurochem 138: 145-162. doi: 10.1111/jnc.13623
- Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth AJ, Kaspar BK, Meyer K, Shaw PJ, Grierson AJ, De Vos KJ (2016). The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO Jl 35(15): 1656-1676. doi: 10.15252/embj.201694401
- Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb Cheynel I, Oulad Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, Charlet Berguerand N (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J 35(12): 1276-1297. doi: 10.15252/embj.201593350
- Ji YJ, Ugolino J, Brady NR, Hamacher-Brady A, Wang J (2017). Systemic deregulation of autophagy upon loss of ALS- and FTD-linked C9orf72. Autophagy 13(7): 1254–1255. doi: 10.1080/15548627.2017.1299312
- Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, Charlet-Berguerand N (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J 35(12): 1276-1297. doi: 10.15252/embj.201593350
- Zhang YY, Burberry A, Wang JY, Sandoe J, Ghosh S, Udeshi ND, Svinkina T, Mordes DA, Mok J, Charlton M, Li QZ, Carr SA, Eggan K (2018). The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes Dev 32(13-14): 929-943. doi: 10.1101/gad.313932.118
- Seto S, Sugaya K, Tsujimura K, Nagata T, Horii T, Koide Y (2013). Rab39a Interacts with Phosphatidylinositol 3-Kinase and Negatively Regulates Autophagy Induced by Lipopolysaccharide Stimulation in Macrophages. Plos One 8(12): e83324. doi: 10.1371/journal.pone.0083324
- Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, Hu F (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun 4(1): 51. doi: 10.1186/s40478-016-0324-5
- Lee J-A, Beigneux A, Ahmad ST, Young SG, Gao F-B (2007). ESCRT-III Dysfunction Causes Autophagosome Accumulation and Neurodegeneration. Curr Biol 17(18): 1561-1567. doi: 10.1016/j.cub.2007.07.029
- Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerød L, Fisher EMC, Isaacs A, Brech A, Stenmark H, Simonsen A (2007). Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179(3): 485-500. doi: 10.1083/jcb.200702115
- Lefebvre C, Legouis R, Culetto E (2018). ESCRT and autophagies: Endosomal functions and beyond. Semin Cell Dev Biol 74: 21-28. doi: 10.1016/j.semcdb.2017.08.014
- Chang MC, Srinivasan K, Friedman BA, Suto E, Modrusan Z, Lee WP, Kaminker JS, Hansen DV, Sheng M (2017). Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J Exp Med 214(9): 2611-2628. doi: 10.1084/jem.20160999
- Ser A, Vollrath JT, Sechi A, Johann S, Roos A, Yamoah A, Katona I, Bohlega S, Wiemuth D, Tian Y, Schmidt A, Vervoorts J, Dohmen M, Beyer C, Anink J, Aronica E, Troost D, Weis J, Goswami A (2017). The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ 24(10): 1655-1671. doi: 10.1038/cdd.2017.88
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurro MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G, et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68(5): 857-864. doi: 10.1016/j.neuron.2010.11.036
- Watts GDJ, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36(4): 377-381. doi: 10.1038/ng1332
- Johnson AE, Shu H, Hauswirth AG, Tong A, Davis GW (2015). VCP-dependent muscle degeneration is linked to defects in a dynamic tubular lysosomal network in vivo. eLife 4: e07366. doi: 10.7554/eLife.07366
- Kustermann M, Manta L, Paone C, Kustermann J, Lausser L, Wiesner C, Eichinger L, Clemen CS, Schröder R, Kestler HA, Sandri M, Rottbauer W, Just S (2018). Loss of the novel Vcp (valosin containing protein) interactor Washc4 interferes with autophagy-mediated proteostasis in striated muscle and leads to myopathy in vivo. Autophagy 14(11): 1911–1927. doi: 10.1080/15548627.2018.1491491
- Ritson GP, Custer SK, Freibaum BD, Guinto JB, Geffel D, Moore J, Tang W, Winton MJ, Neumann M, Trojanowski JQ, Lee VMY, Forman MS, Taylor JP (2010). TDP-43 Mediates Degeneration in a Novel Drosophila Model of Disease Caused by Mutations in VCP/p97. J Neurosci 30(22): 7729-7739. doi: 10.1523/jneurosci.5894-09.2010
- Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, Shen CKJ (2012). Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A 109(37): 15024-15029. doi: 10.1073/pnas.1206362109
- Wang Y, Chen S, Wang Z, Zhang X, Yang D, Zhang X, Li L, Le W (2011). Rapamycin treatment augments motor neuron degeneration in SOD1G93A mouse model of amyotrophic lateral sclerosis. Autophagy 7(4): 412-425. PMID: 21193837
- Feng HL, Leng Y, Ma CH, Zhang J, Ren M, Chuang DM (2008). Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 155(3): 567-572. doi: 10.1016/j.neuroscience.2008.06.040
- Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A, Bellio N, Lenzi P, Modugno N, Siciliano G, Isidoro C, Murri L, Ruggieri S, Paparelli A (2008). Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 105(6): 2052-2057. doi: 10.1073/pnas.0708022105
- Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, Court FA, van Zundert B, Hetz C (2013). Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9(9): 1308-1320. doi: 10.4161/auto.25188
- Chang C-F, Lee Y-C, Lee K-H, Lin H-C, Chen C-L, Shen C-KJ, Huang C-C (2016). Therapeutic effect of berberine on TDP-43-related pathogenesis in FTLD and ALS. J Biomed Sci 23(1): 72-72. doi: 10.1186/s12929-016-0290-z
- Sun J, Mu Y, Jiang Y, Song R, Yi J, Zhou J, Sun J, Jiao X, Prinz RA, Li Y, Xu X (2018). Inhibition of p70 S6 kinase activity by A77 1726 induces autophagy and enhances the degradation of superoxide dismutase 1 (SOD1) protein aggregates. Cell Death Dis 9(3): 407-407. doi: 10.1038/s41419-018-0441-0
- Zhou Q-M, Zhang J-J, Li S, Chen S, Le W-D (2017). n-butylidenephthalide treatment prolongs life span and attenuates motor neuron loss in SOD1G93A mouse model of amyotrophic lateral sclerosis. CNS Neurosci Ther 23(5): 375-385. doi: 10.1111/cns.12681
ACKNOWLEDGMENTS
This work was supported by the NSFC (no.91754115, no.31771531), supported by Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme(GDUPS), and supported by the Science and Technology Planning Project of Guangdong Province (2017B090901051, 2016A020215152) to Du Feng.
COPYRIGHT
© 2019

Recent progress in the role of autophagy in neurological diseases by Meng et al. is licensed under a Creative Commons Attribution 4.0 International License.


