Reviews:
Cell Stress, Vol. 2, No. 7, pp. 162 - 175; doi: 10.15698/cst2018.07.144
Foie gras and liver regeneration: a fat dilemma

1 Department of Experimental Medicine, University of Perugia, Perugia, Italy.
Keywords: liver regeneration, Fatty Liver, ER-stress, NAFLD, SIRT1, Lipid metabolism, steatosis.
Abbreviations:
ER - Endoplasmic Reticulum, ERAD - ER-associated degradation,
NAFLD - non-alcoholic fatty liver disease,
NASH - non-alcoholic steatohepatitis,
PH - partial hepatectomy,
UPR - unfolded protein response.
Received originally: 21/11/2017 Received in revised form: 18/05/2018
Accepted: 22/05/2018
Published: 14/06/2018
Correspondence:
M.A. Della Fazia, Department of Experimental Medicine, University of Perugia, piazza Severi 1, 06132 Perugia, Italy. Tel: 075 585 8111 mariaagnese.dellafazia@unipg.it
G. Servillo, Department of Experimental Medicine, University of Perugia, piazza Severi 1, 06132 Perugia, Italy. Tel: 075 585 8110 giuseppe.servillo@unipg.it
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Maria Agnese Della Fazia and Giuseppe Servillo (2018). Foie gras and liver regeneration: a fat dilemma. Cell Stress 2(7): 162-175. doi: 10.15698/cst2018.07.144
Abstract
The liver has a unique ability of regenerating after injuries or partial loss of its mass. The mechanisms responsible for liver regeneration – mostly occurring when the hepatic tissue is damaged or functionally compromised by metabolic stress – have been studied in considerable detail over the last few decades, because this phenomenon has both basic-biology and clinical relevance. More specifically, recent interest has been focusing on the widespread occurrence of abnormal nutritional habits in the Western world that result in an increased prevalence of non-alcoholic fatty liver disease (NAFLD). NAFLD is closely associated with insulin resistance and dyslipidemia, and it represents a major clinical challenge. The disease may progress to steatohepatitis with persistent inflammation and progressive liver damage, both of which will compromise regeneration under conditions of partial hepatectomy in surgical oncology or in liver transplantation procedures. Here, we analyze the impact of ER stress and SIRT1 in lipid metabolism and in fatty liver pathology, and their consequences on liver regeneration. Moreover, we discuss the fine interplay between ER stress and SIRT1 functioning when contextualized to liver regeneration. An improved understanding of the cellular and molecular intricacies contributing to liver regeneration could be of great clinical relevance in areas as diverse as obesity, metabolic syndrome and type 2 diabetes, as well as oncology and transplantation.
INTRODUCTION
The liver has a unique ability to recover its mass after parenchymal tissue loss, a phenomenon known as liver regeneration [1][2][3]. Traditionally, not only does liver regeneration represent an experimental means of elucidating the basic biology of hepatocyte proliferation, but it is also important from a clinical perspective, in that the liver can be injured by a variety of different noxae, including metabolic diseases, infections, toxin-related pathologies, and autoimmunity [4][5][6]. Liver regeneration is likewise important in surgery, as partial hepatectomy (PH) can be performed as a means of treating hepatocellular carcinoma, and it is used in liver transplantation from live donors as well [7][8][9].
–
A dramatic increase in hepatic steatosis is being observed over the past few years. Non-alcoholic fatty liver disease (NAFLD) – a liver pathology closely associated with insulin resistance and the dyslipidemia-metabolic syndrome [10][11][12] – is present in a percentage as high as approximately 20–30% of any apparently healthy western populations, thus representing a clinical challenge worldwide [13]. Moreover, a significant percentage of individuals with NAFLD will progress to non-alcoholic steatohepatitis (NASH) [14][15][16]. Considering that as many as 25% patients with NASH will develop cirrhosis, it can be assumed that about 2% of people currently with NAFLD are expected to progress to cirrhosis [12][17]. Moreover, patients with NAFLD have an increased risk of developing hepatocellular carcinoma (HCC) [18]. Of note, in liver transplantation, macroscopic steatosis is associated with a higher risk of graft malfunctioning in the recipient. Because of so high a percentage of people with NAFLD, many potential donors are not eligible for donation [9].
–
Studies in rodents and humans have demonstrated an altered liver-regeneration pattern associated with fatty liver diseases [19][20][21][22]. In all of those pathological conditions, an improved understanding of the molecular mechanisms underlying liver regeneration would be of great value from both a basic biology and clinical points of view. In this review, we will focus on the pathophysiology of fat accumulation in the liver and its consequences on liver regeneration as well as liver diseases of major relevance.
LIVER REGENERATION IN EXPERIMENTAL MODELS
The liver presents two specific peculiarities in that it (a) maintains the homeostasis of all of the most important metabolic pathways (as regards lipids, carbohydrates and proteins) in the body [23][24][25][26] and (b) will reconstitute the original hepatic mass after injuries or partial removal of its parenchymal tissue. The two properties are interrelated, and any anomalies in metabolic homeostasis are reflected in altered liver-regeneration patterns. The capacity of the residual hepatocytes to proliferate after PH has been widely used as an experimental model of hepatocyte proliferation in vivo [1][2][3][27]. Two important aspects of liver regeneration have been considered in detail, namely, (a) the proliferative wave whereby stable cells begin to replicate in a synchronous way, and (b) the proliferative arrest occurring when replicating cells have reconstituted the original mass.
–
Since the first scientific report on liver regeneration one hundred years ago [28], many efforts have been made to clarify the underlying signals and mechanisms, both cellular and molecular in nature. Over the years, many molecules have been credited with an important role in liver regeneration, but none of them have been proven to represent, singly, the pivotal factor in liver mass reconstitution [29][30][31][32]. With the advent of genetically deficient knockout (KO) mice, several molecular pathways associated with those molecules have been identified as critically intercrossing at the interface of the proliferative wave and the subsequent proliferative arrest during regeneration. More often than not, the lack of a single – albeit relevant – gene in KO mice was only found to delay the process of liver regeneration, pointing to the mechanistic intricacies, and perhaps the redundancy thereof, whereby many molecules and pathways contribute to liver reconstitution [33][34][35].
–
Nelson Fausto proposed to assign molecules and mechanisms involved in liver regeneration to three major categories, namely, cytokines, growth factors and metabolic networks. In the process of liver regeneration, a variety of genes need to modify their expressions. An early set of genes is required to initiate proliferation in otherwise stable cells, which are thus forced to duplicate [36]. An initial “priming phase” is, in fact, driven primarily by cytokines, such as IL-6 and TNFα [37], and cAMP [32]. By recruiting NF-κB, STAT3 and PKA, a wide number of genes become transcriptionally activated and coordinately act on residual hepatocytes [36], so to result – in turn – in transcriptional activation of the so-called early genes (such as c-fos, c-myc, and c-jun), which initiate the actual process of liver regeneration [38][39]. Although those factors and factor-encoding genes involved in priming were previously thought as being essential to the regeneration process, studies in KO mice with PH have later shown that their respective genetic deficiencies cause only a delay in growth kinetic patterns, rather than a block in cell proliferation or an increased mortality. Therefore, the many factors associated with the priming phase do contribute to regeneration but are dispensable for overall successful completion of the process over the longer term.
–
The priming phase that primes residual hepatocytes for subsequent proliferation requires that growth factors trigger the transition from the G1 to the S phase in hepatocytes. Increased levels of hepatocyte growth factor (HGF) and epidermal growth factor (EGF) – together with the activation of the respective receptors – have long been known to be a prerequisite for proliferation [40][41][42][43]. However, a variety of additional growth factors are now known to be crucially involved in this initial process [44]. Remarkably, despite these changes in transcriptional programs, residual hepatocytes maintain an ability to meet the basic metabolic requirements of the whole body (in terms of glucose, amino-acid, and lipid metabolism). Even when as little as 30% (or even less) of the initial mass remains functional, metabolic homeostasis is not compromised to the benefit of the newly started proliferative process.
–
Notably, the many studies on liver regeneration are not to be taken as a merely mechanistic analysis of how stable cells are forced to expand; rather, they represent an extremely useful model to gain insight into basic biology issues [45][46][47][48][49] as well into the pathophysiology of liver functioning, with general regard to hepatology, transplantation, and hepatocellular carcinoma treatment [9]. Many of those studies on liver regeneration have been making use of genetically deficient KO mice, with the aim of dissecting the contributions of individual genes to the regeneration process. It was only recently that one – perhaps, more clinically relevant – approach has come of age as a way to expand upon the previous information, namely, the analysis of the different patterns of liver regenerations in mice with specific dietary restrictions, such as low- or high-fat diets, low- or high-carbohydrate diets, and diets with low or high protein content [50][51].
–
Many questions still remain unanswered, mainly as to the dynamics of liver regeneration [21][52]. Among those, one of the most crucial is the functional significance of the transient steatosis observed in residual hepatocytes after PH [53][54][55][56]. The issue has been widely investigated in a variety of experimental models. Over the first few hours of PH, the liver accumulates lipids, an event that is indispensable to successful liver regeneration [19][57]. The major lipids being stored are free fatty acids, which are mostly mobilized from adipose tissue, with only a minority of them being derived from hepatic synthesis [58][59][60]. In most studies, those lipids have been identified as being the “fuel” necessary to ignite liver regeneration [61][62][63]. Additional studies – using blockade of fat accumulation either by drugs or in specific KO mice – have indicated that not only does the transient steatosis fuel the process, but it also drives the correct modality of regeneration [22]. Yet, the precise mechanisms governing such a complex phenomenon have been unclear. Perhaps one consideration holds true: after PH, reconstitution of the liver mass has a priority, and the accumulated lipids need to be utilized as a source of energy to guarantee proper regeneration [64] (Fig. 1).
–
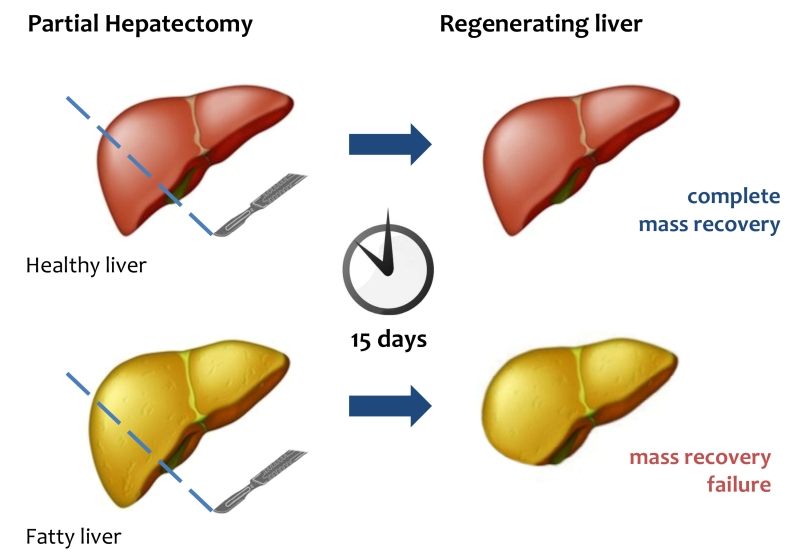 | FIGURE 1: Liver regeneration failure in fatty liver. After PH, healthy liver can completely restore its mass and functional activity within 15 days. In fatty liver mass and functional recovery is compromised. |
LIPID METABOLISM IN LIVER AND ER STRESS
The liver has an essential role in controlling lipid metabolism in the whole body. Cholesterol and fatty acids can be also synthesized and regulated in the liver, which coordinates the assembly of lipoproteins, mainly LDL (low density lipoprotein) and VLDL (very low density lipoprotein) [23][24][65]. Under normal conditions, an overall balance between input and output of lipids in liver is maintained. Changes in this balance predispose to specific diseases such as fatty liver disease.
–
An important role in lipid metabolism is played by the Endoplasmic Reticulum (ER) [66][67]. Several enzymes and proteins involved in lipid metabolism are present in the ER [68][69][70]. Many physio-pathologic conditions will alter ER homeostasis and induce ER stress [71][72][73]. A rapid request for secretory or membrane protein synthesis, as it occurs in liver regeneration, or alterations in redox or Ca2+ homeostasis, are conditions whereby the ER is subjected to high levels of stress [74][75]. In response to many stressors, the ER triggers a well-defined process that causes the cell either to restore normal functioning or to undergo apoptosis. To accomplish the former outcome – and circumvent any potentially dangerous effects of unfolded or misfolded proteins – the ER resorts to a system of protein-quality control referred to as ER-associated degradation or ERAD [76][77][78]. If ERAD, however, fails to restore protein homeostasis, the ER activates a more complex system, namely, the unfolded protein response (UPR) [79][80][81]. The UPR system comprises three pathways involving transcriptional or translational regulators aimed at normalizing ER function. UPR represents a major stress pathway controlled by the chaperone 78-kDa glucose-regulated protein (GRP78) to mediate IRE1, PERK, and ATF6 signaling [82][83][84][85]. Those three proteins – namely, PKR-like ER kinase (PERK), inositol-requiring enzyme-1 (IRE1), and cyclic-AMP-dependent transcription factor ATF-6α (ATF6) – are, under normal conditions, linked to the ER-chaperone GRP78. Upon ER-stress induction, GRP78 binds the unfolded or misfolded proteins as they are released in the ER lumen, thus enabling activation of the three transmembrane proteins involved in UPR. When activated, PERK homodimerizes and, in turn, phosphorylates the eukaryotic initiation factor 2α (eIF2α), which prevents the 80S ribosome assembly, leading to the arrest of protein synthesis [82][86].
–
Even though phosphorylation of eIF2α arrests protein synthesis it allows the translation of selected protein including the transcription factor ATF4, which is overexpressed during ER stress and regulates expression of a number of genes responsible for amino-acid metabolism and apoptosis, including C/EBP homologous protein (CHOP) [87]. Notably, ATF4 controls the expression of GADD34, which in turn plays an important role in dephosphorylating eIF2α, thus activating a key feedback mechanism to restore protein synthesis [88]. Of note, eIF2a can be phosphorylated not only via activation of the UPR system, but also by other kinases involved in the stress response [89].
–
The ER stress-driven UPR system makes use of another transmembrane protein, IRE1. IRE1 is a multifunctional protein, capable of kinase and ribonuclease activities [90]. The RNAse activity allows the generation of a splicing form of the X-box-binding protein 1 (XBP1), which acquires an ability to transcribe ER-chaperons, proteins of the ERAD system, and proteins involved in fatty-acid metabolism [91][92][93].
–
ATF6 is a further UPR-pathway protein activated by ER stress. ATF6 binds GRP78 in the ER, yet in response to stressors, it is released from GRP78 and migrates to the Golgi, where it is cleaved and moves to the nucleus. ATF6 works as a transcription factor coordinating the expression of different components of the UPR system (i.e., GRP78), ER-chaperones, XBP1, the pro-apoptotic factor CHOP, as well as other components of ER-stress response [94][95].
–
Finally, the cell to tackle against stressors has developed, ERAD, UPR, or autophagy, but in the case of failure in adaptive response the UPR sensors addressed the cell towards apoptosis [96] (Fig. 2).
–
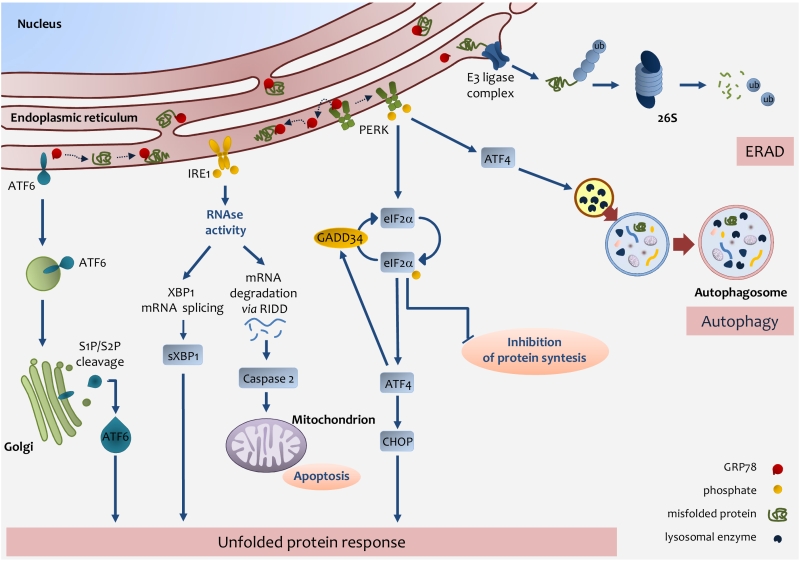 |
FIGURE 2: Endoplasmic reticulum (ER) stress pathways. ER stress occurs through the accu-mulation of misfolded proteins in the ER lumen. The role of the UPR is to re-establish ER homeostasis by reducing de novo protein synthesis and improving the folding and clearance capacity of the ER. The first mechanism against ER stress is ERAD, by which misfolded proteins are translocated back to the cytosol, ubiquitylated and degraded by the proteasome. When the level of misfolded proteins is too high, the cell activates UPR consisting in the activation of three transmembrane pathways. The main effectors are PERK, IRE1 and ATF6. Effectors activation is initiated by the removal of GRP78 allowing the translocation of the latter from the ER membrane to the ER lumen where it associates with unfolded proteins. eIF2α phosphorylation inhibits protein translation except for ATF4 which induces the expression of factors involved in antioxidant defence, amino acid metabolism, autophagy and apoptosis, such as CHOP. ATF4 also induces the expression of GADD34, which expression enables the dephosphorylation of eIF2α and the re-initiation of translation. PERK-mediated induction of ATF4, can also promote the expression of key autophagy-related proteins, which are needed for autophagosome formation. Once activated, IRE1 promotes the splicing of XBP1 mRNA, which is then translated into the active sXBP1, which transactivates the expression of components related to protein folding, ERAD and protein quality control. IRE1 also promotes the degradation of RNAs localized in the ER vicinity by regulated IRE1-dependent decay (RIDD), which induces caspase-2 and mitochondrial apoptosis. Upon GRP78 release, ATF6 is transferred to the Golgi apparatus as inactive precursors and cleaved by membrane-bound site-1 (S1P) and site-2 (S2P) proteases into an active form, which induces the expression of chaperones and UPR components. IRE1-mediated activation of XBP1 as well as ATF6 activation induces the expression of chaperones and UPR components. |
ER STRESS AND LIVER REGENERATION
Because of the role of liver in controlling lipid homeostasis, it is clear that the UPR system is considered to be pivotal in the response to a number of stressors acting on hepatocytes. Chemical or naturally occurring toxicants act on hepatocytes and changes in cellular status – such as nutritional and proliferative changes or rapid functional requests – are accomplished via transient ER stress and UPR activation [97][98][99][100]. Indeed, proliferation of residual hepatocytes in the onset of liver regeneration needs abundant protein synthesis, which increases protein folding demand within the ER. At present, UPR function is considered a major component of liver regeneration. The analysis of pivotal players in UPR shows a triggering event during liver regeneration. Physiological response involves a rapid increase and activation of ER-stress genes after PH. IRE1a pathway, PERK, pIF2a and CHOP increase their expression in the first hours after partial hepatectomy [101][102][103].In particular, after a 70% PH, residual hepatocytes undergo changes that drive ER stress [20][104]. As a matter of fact, the residual tissue (30%) needs to preserve the overall homeostasis in the body while engaged in reconstituting liver mass. The latter event requires that residual hepatocytes are able to synthesize and assemble all the cellular components needed for regeneration, which calls for the onset of a transient ER-stress status, the hallmark of which is temporary steatosis, reversed only by the time of regeneration completion [20][53][64].
–
Such a bidirectional relationship would imply that an excess of dietary lipid intake resulting in steatosis might determine a status of chronic activation of the ER-stress response. Experiments in obese rodents have indeed provided evidence for the persistent activation of the UPR system, a condition shared clinically by patients with severe steatosis, NAFLD, or NASH [105][106][107]. The major mechanism responsible for the association between steatosis and activation of the UPR system seems to be related to changes in membrane fluidity and consequent loss of Ca2+ homeostasis. Changes in membrane composition and fluidity have been described in obese mice that display an altered ratio between the two main membrane phospholipids, phosphatidylcholine and phosphatidylethanolamine [108]. An altered ratio between the two phospholipids leads to ER membrane modification, which, in turn, affects Ca2+ homeostasis. As demonstrated by a series of elegant experiments, a decrease in Ca2+ causes remarkable stress in the ER via reduction in the Ca2+-dependent chaperone component. Overall, these data substantiate the role of lipids as one of the most important ER stressor [109][110].
–
Many authors have identified ER stress as being, itself, the primary cause of the de novo synthesis of lipids either under conditions of steatosis-associated insulin resistance or of chronic ER stress-induced proteolytic cleavage of SREBP-1c, which translocates to the nucleus and activates a cohort of genes involved in lipogenesis [111]. Therefore, the outcome of chronic activation of ER stress by lipids is steatosis and subsequent onset of NAFLD. This is determined by lipid accumulation in the liver and ER-stress activation, which plays an important role per se in the pathogenesis of NAFLD, mainly by direct control of de novo lipogenesis and by mitochondria modifications [112][113]. On the long term, chronic ER stress recruits additional mechanisms in the liver, including a reduction in VLDL secretion or a change in the insulin response, which exacerbates the condition [114]. Such a prolonged condition of the liver causes increased oxidative stress accompanied by inflammation and apoptosis, eventually resulting in the onset of NASH. Overall, chronic activation of ER stress is a pathological condition that determines a severe impairment in liver functioning [104][115].
–
The regenerative ability of the liver under ER stress is altered and even in absence of severe steatosis or NAFLD, the residual hepatocytes have a delay in their ability to proliferate properly [116][117][118]. Recent studies in mice fed a high-fat diet for 10 weeks to induce steatosis showed that the classical genes involved in ER stress – with the exclusion of sXBP-1 – were not overexpressed. After PH, a delay in the proliferation of residual hepatocytes was observed in mice fed the high-fat diet; higher a ctivation of ER-stress genes, such as GRP78, IRE1a, ATF6, PERK, sXBP-1 and CHOP, was detected [20]. Moreover, persistent fat accumulation was present in mice fed the diet. Mice genetically deficient for the 3 β-hydroxysterol Δ14-reductase (TM7SF2)-encoding gene (TM7SF2 KO mice, which lack the ER enzyme involved in cholesterol synthesis) showed abnormal activation of the ER-stress response. PH in TM7SF2 KO mice resulted in an impaired proliferation relative to wild-type counterparts, with a delay in the cell-cycle G1/S transition and early activation of GRP78. Furthermore, the KO mice accumulated an anomalous amount of hepatic triglycerides until 60 hours of PH. Unusual activation of p53 and persistently elevated levels of p21 were observed in the KO mice during liver regeneration. Because of the control of p21 on CHOP, it is conceivable that elevated level of p21 in those mice is related to the activation of the ER-stress response by CHOP [104]. The analysis of liver regeneration after PH in different KO mice, in which any genes involved in ER-stress are deleted, shows regeneration abnormalities with altered proliferation of the residual hepatocytes. Impairment in regenerative process has been detected in mice with IRE1a hepatocyte-specific deletion [101]. Moreover, the ER stress has been defined as an important cellular process in liver regeneration under Ischemia/Reperfusion Injury [119] as demonstrated in CHOP KO mice [120].
–
Whether a perturbation in the ER-stress response is directly responsible for the delay in liver regeneration, or an altered ER-stress response sustains fat accumulation responsible for the impaired liver regeneration, is still to be defined.
SIRTUINS AND LIPID METABOLISM
Many studies have been describing an important role for sirtuins in the metabolism of liver lipids. Sirtuins are a family of proteins that share a number of functions and which conserve NAD+-dependent histone and protein deacetylase functions. In mammals there are seven sirtuins with different cellular functions ranging from energy metabolism, cellular stress resistance, genomic stability to aging and tumorigenesis [121][122]. Each Sirtuin has distinct functions and subcellular localizations. SIRT1 and SIRT2 are in both the nucleus and the cytosol, SIRT3, SIRT4, and SIRT5 are in the mitochondria and SIRT6 and SIRT7 are localized in the nucleus [123]. Among them, SIRT1, SIRT3, SIRT6 and SIRT7 have been discovered as involved in fat liver metabolism [124][125][126][127][128].
–
The silent information regulator 1 (SIRT1) represents one of the best-characterized members of the mammalian sirtuin family of NAD-dependent histone deacetylases. SIRT1 is a nutrient sensor and has a crucial role in the control of ageing in different organisms [129]. Moreover, SIRT1 has a pivotal role in the control of normal liver function in mammals, participating in the regulation of metabolic processes such as gluconeogenesis, fatty acid β-oxidation and cholesterol flux [122].
–
Of particular interest, SIRT1 has been known as an important regulator of circadian rhythms [130][131][132][133]. SIRT1 directs circadian oscillation in hepatocytes through rhythmic deacetylation of histone H3 at the promoter of clock-controlled genes. Moreover, SIRT1 controls deacetylation of important circadian regulators such as BMAL1 and PER2 [130][132]. Deficiency of liver-specific SIRT1 – using SIRT1-KO (SirtLKO) mice – as well as pharmacological modulation of SIRT1 expression lead to major changes in hepatic circadian gene expression and lipid metabolism [134][135].
–
SIRT1 crucially acts as a sensor in metabolic and energy control of the cell [136][137]. SIRT1 activity is mediated by NAD+ levels [138]. Because the liver is the major organ controlling homeostasis, it is clear that perturbations driven by high-fat or high-calorie diets, alcohol or drugs altering NAD+ levels will all affect SIRT1 transcriptional activity [124][139][140][141]. Several participants in liver homeostasis are regulated by SIRT1, most of them being transcription factors with a role in lipid metabolism, such as the peroxisome proliferator-activated receptor-α (PPARα), peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) [142], carbohydrate response element binding protein (ChREBP), sterol regulatory element binding protein-1c (SREBP-1) [143], and NF-κB [144][145]. The control of SIRT1 occurs de facto via deacetylation of transcription factors involved in lipid metabolism [146]. SREBP-1c and ChREBP deacetylation is accompanied by an arrested transcription of downstream genes, leading to lipogenesis. PPARα and PGC-1α deacetylation promotes FA-β-oxidation. Owing to the role of SIRT1 in controlling liver lipid metabolism, many studies have been conducted in SirtLKO mice [147]. In particular, it was found that SirtLKO mice, fed a normal diet, develop enhanced fat accumulation in their livers over their entire lifespans, indicating a clear participation of SIRT1 in the onset of hepatic steatosis [147][148][149][150]. Indeed, more than with steatosis alone, the overall clinical picture is consistent with an underlying inflammatory condition and increased ROS production, both of which accompany the hyperglycemia and insulin resistance typically driven by steatosis in SirtLKO mice [151].
–
Interestingly, PH performed in SirtLKO mice results in impaired liver regeneration, with a clear delay in the G1/S transition, deregulation of cyclins and related CDKs and delayed regeneration. Moreover, the regenerating liver from SirtLKO mice shows unusual accumulation of lipids, with increased levels of TAGs (triacylglycerol), NEFA (non-esterified fatty acid) and cholesterol, thus confirming the crucial role of SIRT1 in regulating fat in liver under regeneration. Notably, under the same conditions, liver regeneration is characterized by downregulation of PPARα-related genes, thus substantiating a role for SIRT1 in controlling PPARα and, in turn, fatty acid β-oxidation [152]. These findings are in agreement with previous results in PPARα KO mice, in which a delay occurs in liver regeneration relative to control counterparts. Overall it is clear that, by balancing fat composition, SIRT1 has an important role in liver regeneration [153].
–
The recent finding of an interaction between SIRT1 and ER stress in the liver has been receiving much attention [154]. Whether lack of SIRT1 after PH – leading to hepatic fat composition – drives ER-stress activation or, conversely, it is the lack of SIRT1 – resulting in unrestrained ER stress – that matters so much, remains to be explained. On the one hand, many studies have demonstrated that SIRT1 overexpression alleviates ER stress; SIRT1 overexpression in obese mice is accompanied by a reduction in ER stress and steatosis [155][156][157]. On the other hand, recent studies have credited SIRT1 with an ability to control the activation of ER stress, by deacetyling the fundamental transcription factor XBP1, and by controlling eIF2α, which triggers ER stress [158]. Overall, the interplay between ER stress and SIRT1 is intricate and complex, but recent reports have been providing new insight into this relationship, mostly by focusing on the role of ER stress in controlling SIRT1. In vivo and in vitro experiments showed that, by activating the PI3K-AKT-GSK3b pathway, ER stress would induce expression of SIRT1 [159].
–
Interestingly, SIRT1 and ER stress share another important function in the liver. SIRT1 is a pivotal regulator of circadian rhythms in the liver, where it has an ability to deacetylate histone H3 in a rhythmic manner, which, in turn, controls the promoters of clock-controlled genes as well as BMAL1 and PER2 [130][132]. Altered SIRT1 activity in the liver is reflected in an altered expression of circadian genes and lipid metabolism [134][135]. Importantly, the circadian clock coordinates the activation of important genes regulating ER stress in the liver; an altered circadian rhythm perturbs ER stress and, consequently, lipid metabolism [160][161][162]. Increasing evidence is available regarding the relationship between lipid metabolism and circadian clock, with a focus on timing of food intake, liver stress and liver pathology (Fig. 3).
–
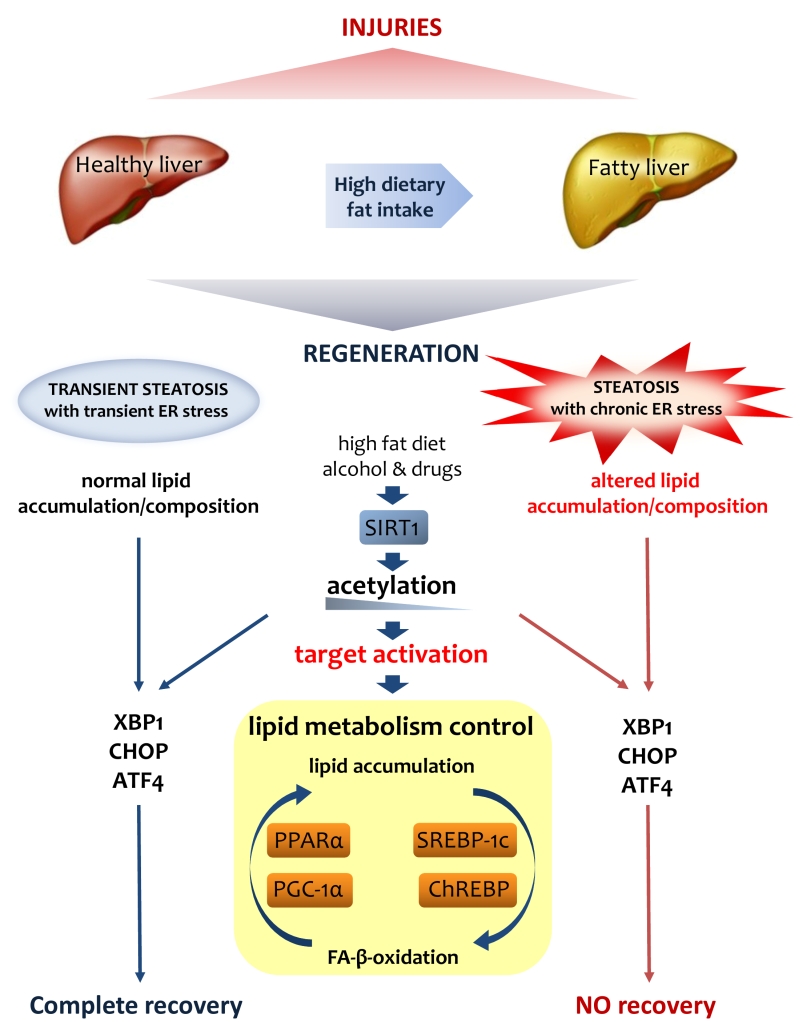 | FIGURE 3: Lipid metabolism is crucial for liver regeneration. Fatty liver has an altered metabolism in lipids that affects its ability to regenerate after various injuries. Lipid accumulation during liver regeneration induces a transient steatosis with transient ER stress that is functional to proper proliferation of hepatocytes. On the contrary, the chronic steatosis and ER stress in fatty liver result in an altered lipid asset that could be addressed as the origin of the failure of regeneration. SIRT1, as a major player in lipid metabolism control, through its deacetylase activity on H3-histone at gene promoters, controls the activation of various UPR genes such as XPB1, CHOP and ATF4. |
–
SIRT3 is a mitochondrial protein, which plays an important role in the control of carbohydrate metabolism, ketogenesis, β-oxidation, and amino-acid metabolism [163]. Chronic high fat diet and obesity reduced SIRT3 activity, which in turn is associated with fatty liver. Indeed, analysis of SIRT3-KO mice shows no metabolic difference respect to the WT mice even if differences have been found in the mitochondrial acetylation proteins [164]. Hyperacetylation of mitochondrial protein found in SIRT3-KO mice accelerates the development of metabolic syndrome with steatohepatitis [165]. Recently, a study on pancreatic b-cells showed that SIRT3 is involved in the regulation of ER-stress down-regulating the gene expression of protein of ER-stress [165]. SIRT6 and SIRT7 have been investigated to analyze their role in NAFLD. The analysis of Hepatocyte-Specific SIRT6-KO mice have revealed a predisposition to NAFLD when fed with a high-fat and high-fructose (HFHF) diet for 16 weeks. HFHF-diet induced in SIRT6-KO mice an increased in hepatic steatosis and inflammation with liver fibrosis and oxidative stress [166].
–
SIRT7 is a nuclear protein, highly expressed in the liver that regulates metabolic homeostasis [167]. It has been demonstrated that SIRT7 specific liver deficient mice develop steatosis with elevated expression of inflammatory markers indicating a turning towards NASH. Moreover, it has been revealed that in SIRT7 specific liver deficient mice the liver steatosis is not accompanied with obesity. Notably, it has been discovered that in liver SIRT7 regulates ER-stress by repressing Myc activity [168].
–
Nevertheless, a clear picture of the complex relationship between Sirtuins and ER stress is still lacking (Table 1).
–
TABLE 1. Summary of effects of Non-alcoholic fatty liver disease (NAFLD) in various models on liver regeneration. |
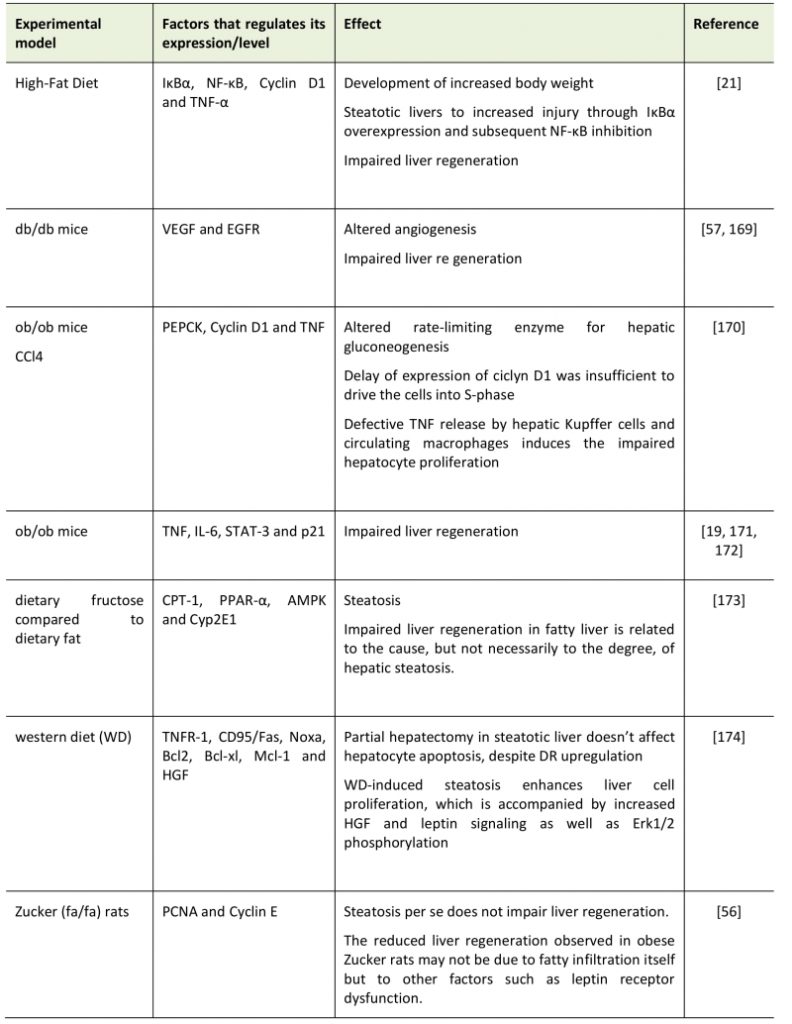 |
| [19][21][56][57][169][170][171][172][173][174] |
CONCLUSIONS
Liver regeneration represents a unique model for studying proliferation under conditions in which stable cells are induced to proliferate to rectify any parenchymal loss. Yet, the model is not simply meant to prove insight into a biological and legendary curiosity – the legend of Prometheus, chained to a rock, where an eagle ate his liver during the day, and the liver was regenerated during the night due to Prometheus’ immortality. The knowledge, indeed, of the unique and complex mechanisms that determine liver regeneration might be of great help to a better understanding of liver pathology and to improved therapeutic and surgical interventions.
–
Broad panoply of events influences the ability of liver to regenerate, and recently fat liver accumulation has been receiving much attention, because of the high incidence of steatosis in humans. Macroscopic or severe steatosis has a negative impact on regeneration of the liver in that it opposes proper regeneration in patients after surgical removal of neoplasia or in liver grafts. Moreover, because of the high incidence of NAFLD, steatosis represents a significant limitation to the availability of potential liver donors.
–
The knowledge of the molecular mechanisms governing liver regeneration and their impact on human liver pathology is becoming increasingly relevant in the clinical context. A better comprehension of the factors responsible for fat accumulation and their potential role in liver regeneration might permit the identification of novel druggable targets in hepatology [169].
References
- N. Fausto, "Liver regeneration.", Journal of hepatology, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10728791
- G.K. Michalopoulos, and M.C. DeFrances, "Liver Regeneration", Science, vol. 276, pp. 60-66, 1997. http://dx.doi.org/10.1126/science.276.5309.60
- R. Taub, "Liver regeneration: from myth to mechanism", Nature Reviews Molecular Cell Biology, vol. 5, pp. 836-847, 2004. http://dx.doi.org/10.1038/nrm1489
- M. Preziosi, and S. Monga, "Update on the Mechanisms of Liver Regeneration", Seminars in Liver Disease, vol. 37, pp. 141-151, 2017. http://dx.doi.org/10.1055/s-0037-1601351
- G.K. Michalopoulos, "Hepatostat: Liver regeneration and normal liver tissue maintenance", Hepatology, vol. 65, pp. 1384-1392, 2017. http://dx.doi.org/10.1002/hep.28988
- Y.J. Kwon, K.G. Lee, and D. Choi, "Clinical implications of advances in liver regeneration", Clinical and Molecular Hepatology, vol. 21, pp. 7, 2015. http://dx.doi.org/10.3350/cmh.2015.21.1.7
- S.S. Huppert, and K.M. Campbell, "Emerging advancements in liver regeneration and organogenesis as tools for liver replacement", Current Opinion in Organ Transplantation, vol. 21, pp. 581-587, 2016. http://dx.doi.org/10.1097/MOT.0000000000000365
- C.T. Nicolas, R.D. Hickey, H.S. Chen, S.A. Mao, M. Lopera Higuita, Y. Wang, and S.L. Nyberg, "Concise Review: Liver Regenerative Medicine: From Hepatocyte Transplantation to Bioartificial Livers and Bioengineered Grafts", Stem Cells, vol. 35, pp. 42-50, 2016. http://dx.doi.org/10.1002/stem.2500
- S.J. Forbes, and P.N. Newsome, "Liver regeneration — mechanisms and models to clinical application", Nature Reviews Gastroenterology & Hepatology, vol. 13, pp. 473-485, 2016. http://dx.doi.org/10.1038/nrgastro.2016.97
- E.K. Speliotes, J.M. Massaro, U. Hoffmann, R.S. Vasan, J.B. Meigs, D.V. Sahani, J.N. Hirschhorn, C.J. O'Donnell, and C.S. Fox, "Fatty Liver Is Associated With Dyslipidemia and Dysglycemia Independent of Visceral Fat: the Framingham Heart Study", Hepatology, vol. 51, pp. 1979-1987, 2010. http://dx.doi.org/10.1002/hep.23593
- L.A. Adams, J.F. Lymp, J. St. Sauver, S.O. Sanderson, K.D. Lindor, A. Feldstein, and P. Angulo, "The Natural History of Nonalcoholic Fatty Liver Disease: A Population-Based Cohort Study", Gastroenterology, vol. 129, pp. 113-121, 2005. http://dx.doi.org/10.1053/j.gastro.2005.04.014
- J.M. Clark, F.L. Brancati, and A.M. Diehl, "Nonalcoholic fatty liver disease.", Gastroenterology, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12016429
- M.V. Machado, and A.M. Diehl, "Pathogenesis of Nonalcoholic Steatohepatitis", Gastroenterology, vol. 150, pp. 1769-1777, 2016. http://dx.doi.org/10.1053/j.gastro.2016.02.066
- A.J. McCullough, "The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease", Clinics in Liver Disease, vol. 8, pp. 521-533, 2004. http://dx.doi.org/10.1016/j.cld.2004.04.004
- J.P. Ong, and Z.M. Younossi, "Epidemiology and Natural History of NAFLD and NASH", Clinics in Liver Disease, vol. 11, pp. 1-16, 2007. http://dx.doi.org/10.1016/j.cld.2007.02.009
- G.A. Michelotti, M.V. Machado, and A.M. Diehl, "NAFLD, NASH and liver cancer", Nature Reviews Gastroenterology & Hepatology, vol. 10, pp. 656-665, 2013. http://dx.doi.org/10.1038/nrgastro.2013.183
- S. Bellentani, G. Saccoccio, F. Masutti, L.S. Crocè, G. Brandi, F. Sasso, G. Cristanini, and C. Tiribelli, "Prevalence of and risk factors for hepatic steatosis in Northern Italy.", Annals of internal medicine, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10644271
- K. Yasui, E. Hashimoto, K. Tokushige, K. Koike, T. Shima, Y. Kanbara, T. Saibara, H. Uto, S. Takami, M. Kawanaka, Y. Komorizono, T. Okanoue, and . , "Clinical and pathological progression of non‐alcoholic steatohepatitis to hepatocellular carcinoma", Hepatology Research, vol. 42, pp. 767-773, 2012. http://dx.doi.org/10.1111/j.1872-034X.2012.00986.x
- S.Q. Yang, H.Z. Lin, A.K. Mandal, J. Huang, and A.M. Diehl, "Disrupted signaling and inhibited regeneration in obese mice with fatty livers: Implications for nonalcoholic fatty liver disease pathophysiology", Hepatology, vol. 34, pp. 694-706, 2001. http://dx.doi.org/10.1053/jhep.2001.28054
- M. Hamano, H. Ezaki, S. Kiso, K. Furuta, M. Egawa, T. Kizu, N. Chatani, Y. Kamada, Y. Yoshida, and T. Takehara, "Lipid overloading during liver regeneration causes delayed hepatocyte DNA replication by increasing ER stress in mice with simple hepatic steatosis", Journal of Gastroenterology, vol. 49, pp. 305-316, 2013. http://dx.doi.org/10.1007/s00535-013-0780-7
- R.A. DeAngelis, M.M. Markiewski, R. Taub, and J.D. Lambris, "A High-Fat Diet Impairs Liver Regeneration in C57BL/6 Mice Through Overexpression of the NF-κB Inhibitor, IκBα * #", Hepatology, vol. 42, pp. 1148-1157, 2005. http://dx.doi.org/10.1002/hep.20879
- E. Shteyer, Y. Liao, L.J. Muglia, P.W. Hruz, and D.A. Rudnick, "Disruption of Hepatic Adipogenesis Is Associated With Impaired Liver Regeneration in Mice", Hepatology, vol. 40, pp. 1322-1332, 2004. http://dx.doi.org/10.1002/hep.20462
- D.L. Silver, "Hepatic Fatty Acid Metabolism and Dysfunction", The Liver, pp. 257-270, 2009. http://dx.doi.org/10.1002/9780470747919.ch18
- D.E. Cohen, "Lipoprotein Metabolism and Cholesterol Balance", The Liver, pp. 271-285, 2009. http://dx.doi.org/10.1002/9780470747919.ch19
- M.H. Oosterveer, and K. Schoonjans, "Hepatic glucose sensing and integrative pathways in the liver", Cellular and Molecular Life Sciences, vol. 71, pp. 1453-1467, 2013. http://dx.doi.org/10.1007/s00018-013-1505-z
- T.J. Wester, G. Kraft, D. Dardevet, S. Polakof, I. Ortigues-Marty, D. Rémond, and I. Savary-Auzeloux, "Nutritional regulation of the anabolic fate of amino acids within the liver in mammals: concepts arising fromin vivostudies", Nutrition Research Reviews, vol. 28, pp. 22-41, 2015. http://dx.doi.org/10.1017/S0954422415000013
- N. Fausto, J.S. Campbell, and K.J. Riehle, "Liver regeneration", Journal of Hepatology, vol. 57, pp. 692-694, 2012. http://dx.doi.org/10.1016/j.jhep.2012.04.016
- B.G. Crawford, "TOXIC JAUNDICE, WITH ATROPHY OF LIVER, FOLLOWED BY REGENERATION AND RECOVERY.", British medical journal, 1918. http://www.ncbi.nlm.nih.gov/pubmed/20769007
- A.M. Diehl, and R.M. Rai, "Liver regeneration 3: Regulation of signal transduction during liver regeneration.", FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 1996. http://www.ncbi.nlm.nih.gov/pubmed/8641555
- R. Taub, "Liver regeneration 4: transcriptional control of liver regeneration.", FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 1996. http://www.ncbi.nlm.nih.gov/pubmed/8647340
- N. Fausto, A.D. Laird, and E.M. Webber, "Liver regeneration. 2. Role of growth factors and cytokines in hepatic regeneration.", FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 1995. http://www.ncbi.nlm.nih.gov/pubmed/8529831
- G. Servillo, M.A. Della Fazia, and P. Sassone-Corsi, "Coupling cAMP Signaling to Transcription in the Liver: Pivotal Role of CREB and CREM", Experimental Cell Research, vol. 275, pp. 143-154, 2002. http://dx.doi.org/10.1006/excr.2002.5491
- D.E. Cressman, L.E. Greenbaum, R.A. DeAngelis, G. Ciliberto, E.E. Furth, V. Poli, and R. Taub, "Liver Failure and Defective Hepatocyte Regeneration in Interleukin-6-Deficient Mice", Science, vol. 274, pp. 1379-1383, 1996. http://dx.doi.org/10.1126/science.274.5291.1379
- Y. Yamada, I. Kirillova, J. Peschon, and N. Fausto, "Initiation of liver growth by tumor necrosis factor: Deficient liver regeneration in mice lacking type I tumor necrosis factor receptor", Proceedings of the National Academy of Sciences, vol. 94, pp. 1441-1446, 1997. http://dx.doi.org/10.1073/pnas.94.4.1441
- G. Servillo, M.A. Della Fazia, and P. Sassone-Corsi, "Transcription factor CREM coordinates the timing of hepatocyte proliferation in the regenerating liver", Genes & Development, vol. 12, pp. 3639-3643, 1998. http://dx.doi.org/10.1101/gad.12.23.3639
- N. Fausto, J.S. Campbell, and K.J. Riehle, "Liver Regeneration", Hepatology, vol. 43, pp. S45-S53, 2006. http://dx.doi.org/10.1002/hep.20969
- M. Iwai, T. Cui, H. Kitamura, M. Saito, and T. Shimazu, "INCREASED SECRETION OF TUMOUR NECROSIS FACTOR AND INTERLEUKIN 6 FROM ISOLATED, PERFUSED LIVER OF RATS AFTER PARTIAL HEPATECTOMY", Cytokine, vol. 13, pp. 60-64, 2001. http://dx.doi.org/10.1006/cyto.2000.0797
- C. Trautwein, T. Rakemann, M. Niehof, S. Rose-John, and M. Manns, "Acute-phase response factor, increased binding, and target gene transcription during liver regeneration", Gastroenterology, vol. 110, pp. 1854-1862, 1996. http://dx.doi.org/10.1053/gast.1996.v110.pm8964411
- A.I. Su, L.G. Guidotti, J.P. Pezacki, F.V. Chisari, and P.G. Schultz, "Gene expression during the priming phase of liver regeneration after partial hepatectomy in mice", Proceedings of the National Academy of Sciences, vol. 99, pp. 11181-11186, 2002. http://dx.doi.org/10.1073/pnas.122359899
- C. Huh, V.M. Factor, A. Sánchez, K. Uchida, E.A. Conner, and S.S. Thorgeirsson, "Hepatocyte growth factor/c-metsignaling pathway is required for efficient liver regeneration and repair", Proceedings of the National Academy of Sciences, vol. 101, pp. 4477-4482, 2004. http://dx.doi.org/10.1073/pnas.0306068101
- M. Borowiak, A.N. Garratt, T. Wüstefeld, M. Strehle, C. Trautwein, and C. Birchmeier, "Met provides essential signals for liver regeneration", Proceedings of the National Academy of Sciences, vol. 101, pp. 10608-10613, 2004. http://dx.doi.org/10.1073/pnas.0403412101
- G.M. Argast, J.S. Campbell, J.T. Brooling, and N. Fausto, "Epidermal Growth Factor Receptor Transactivation Mediates Tumor Necrosis Factor-induced Hepatocyte Replication", Journal of Biological Chemistry, vol. 279, pp. 34530-34536, 2004. http://dx.doi.org/10.1074/jbc.M405703200
- A. Francavilla, P. Ove, L. Polimeno, C. Sciascia, M.L. Coetzee, and T.E. Starzl, "Epidermal growth factor and proliferation in rat hepatocytes in primary culture isolated at different times after partial hepatectomy.", Cancer research, 1986. http://www.ncbi.nlm.nih.gov/pubmed/3002614
- H. Talarmin, C. Rescan, S. Cariou, D. Glaise, G. Zanninelli, M. Bilodeau, P. Loyer, C. Guguen-Guillouzo, and G. Baffet, "The Mitogen-Activated Protein Kinase Kinase/Extracellular Signal-Regulated Kinase Cascade Activation Is a Key Signalling Pathway Involved in the Regulation of G1 Phase Progression in Proliferating Hepatocytes", Molecular and Cellular Biology, vol. 19, pp. 6003-6011, 1999. http://dx.doi.org/10.1128/mcb.19.9.6003
- M.A. Della Fazia, D. Piobbico, D. Bartoli, M. Castelli, S. Brancorsini, M. Viola Magni, and G. Servillo, "lal‐1: a differentially expressed novel gene during proliferation in liver regeneration and in hepatoma cells", Genes to Cells, vol. 7, pp. 1183-1190, 2002. http://dx.doi.org/10.1046/j.1365-2443.2002.00593.x
- M.A.D. Fazia, M. Castelli, D. Bartoli, S. Pieroni, V. Pettirossi, D. Piobbico, M. Viola-Magni, and G. Servillo, "HOPS: a novel cAMP-dependent shuttling protein involved in protein synthesis regulation", Journal of Cell Science, vol. 118, pp. 3185-3194, 2005. http://dx.doi.org/10.1242/jcs.02452
- S. Pieroni, M.A. Della Fazia, M. Castelli, D. Piobbico, D. Bartoli, C. Brunacci, M.M. Bellet, M. Viola-Magni, and G. Servillo, "HOPS is an essential constituent of centrosome assembly", Cell Cycle, vol. 7, pp. 1462-1466, 2008. http://dx.doi.org/10.4161/cc.7.10.5882
- M. Castelli, S. Pieroni, C. Brunacci, D. Piobbico, D. Bartoli, M.M. Bellet, E. Colombo, P.G. Pelicci, M.A. Della Fazia, and G. Servillo, "Hepatocyte odd protein shuttling (HOPS) is a bridging protein in the nucleophosmin-p19Arf network", Oncogene, vol. 32, pp. 3350-3358, 2012. http://dx.doi.org/10.1038/onc.2012.353
- M.M. Bellet, D. Piobbico, D. Bartoli, M. Castelli, S. Pieroni, C. Brunacci, M. Chiacchiaretta, R. Del Sordo, F. Fallarino, A. Sidoni, P. Puccetti, L. Romani, G. Servillo, and M. Agnese Della Fazia, "NEDD4 controls the expression of GUCD1, a protein upregulated in proliferating liver cells", Cell Cycle, vol. 13, pp. 1902-1911, 2014. http://dx.doi.org/10.4161/cc.28760
- H. Chen, Y. Lin, W. Sun, Y. Cai, and H. Li, "Liver Regeneration Is Impaired in Mice with Acute Exposure to a Very Low Carbohydrate Diet", Digestive Diseases and Sciences, vol. 62, pp. 1256-1264, 2017. http://dx.doi.org/10.1007/s10620-017-4519-9
- M. Holeček, "Nutritional modulation of liver regeneration by carbohydrates, lipids, and amino acids: a review", Nutrition, vol. 15, pp. 784-788, 1999. http://dx.doi.org/10.1016/s0899-9007(99)00158-6
- M. Selzner, and P. Clavien, "Failure of regeneration of the steatotic rat liver: disruption at two different levels in the regeneration pathway", Hepatology, vol. 31, pp. 35-42, 2000. http://dx.doi.org/10.1002/hep.510310108
- E. Kachaylo, C. Tschuor, N. Calo, N. Borgeaud, U. Ungethüm, P. Limani, A. Piguet, J. Dufour, M. Foti, R. Graf, P.A. Clavien, and B. Humar, "PTEN Down‐Regulation Promotes β‐Oxidation to Fuel Hypertrophic Liver Growth After Hepatectomy in Mice", Hepatology, vol. 66, pp. 908-921, 2017. http://dx.doi.org/10.1002/hep.29226
- B.H. Zhang, M. Weltman, and G.C. Farrell, "Does steatohepatitis impair liver regeneration? A study in a dietary model of non-alcoholic steatohepatitis in rats.", Journal of gastroenterology and hepatology, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10029293
- M.S. Rao, K. Papreddy, M. Abecassis, and T. Hashimoto, "Regeneration of liver with marked fatty change following partial hepatectomy in rats.", Digestive diseases and sciences, 2001. http://www.ncbi.nlm.nih.gov/pubmed/11575431
- C. Picard, L. Lambotte, P. Starkel, C. Sempoux, A. Saliez, V. Van Den Berge, and Y. Horsmans, "Steatosis is not sufficient to cause an impaired regenerative response after partial hepatectomy in rats", Journal of Hepatology, vol. 36, pp. 645-652, 2002. http://dx.doi.org/10.1016/s0168-8278(02)00038-7
- H. Yamauchi, K. Uetsuka, T. Okada, H. Nakayama, and K. Doi, "Impaired liver regeneration after partial hepatectomy in db/db mice", Experimental and Toxicologic Pathology, vol. 54, pp. 281-286, 2003. http://dx.doi.org/10.1078/0940-2993-00265
- E.P. Newberry, S.M. Kennedy, Y. Xie, J. Luo, S.E. Stanley, C.F. Semenkovich, R.M. Crooke, M.J. Graham, and N.O. Davidson, "Altered hepatic triglyceride content after partial hepatectomy without impaired liver regeneration in multiple murine genetic models†", Hepatology, vol. 48, pp. 1097-1105, 2008. http://dx.doi.org/10.1002/hep.22473
- E.A. Glende, and W.S. Morgan, "Alteration in liver lipid and lipid fatty acid composition after partial hepatectomy in the rat.", Experimental and molecular pathology, 1968. http://www.ncbi.nlm.nih.gov/pubmed/4296682
- C.D. Gove, and D.A. Hems, "Fatty acid synthesis in the regenerating liver of the rat", Biochemical Journal, vol. 170, pp. 1-8, 1978. http://dx.doi.org/10.1042/bj1700001
- T.J. Delahunty, and D. Rubinstein, "Accumulation and release of triglycerides by rat liver following partial hepatectomy.", Journal of lipid research, 1970. http://www.ncbi.nlm.nih.gov/pubmed/5504519
- A.C.M. Camargo, J. Cornicelli, and S.S. Cardoso, "Alteration in Lipid Content of the Liver in the Rat After Partial Hepatectomy.", Experimental Biology and Medicine, vol. 122, pp. 1151-1154, 1966. http://dx.doi.org/10.3181/00379727-122-31349
- G.C. Farrell, "Probing Prometheus: Fat Fueling the Fire?", Hepatology, vol. 40, pp. 1252-1255, 2004. http://dx.doi.org/10.1002/hep.20522
- D.A. Rudnick, and N.O. Davidson, "Functional Relationships between Lipid Metabolism and Liver Regeneration", International Journal of Hepatology, vol. 2012, pp. 1-8, 2012. http://dx.doi.org/10.1155/2012/549241
- A. Canbay, L. Bechmann, and G. Gerken, "Lipid Metabolism in the Liver", Zeitschrift für Gastroenterologie, vol. 45, pp. 35-41, 2007. http://dx.doi.org/10.1055/s-2006-927368
- N. Borgese, M. Francolini, and E. Snapp, "Endoplasmic reticulum architecture: structures in flux", Current Opinion in Cell Biology, vol. 18, pp. 358-364, 2006. http://dx.doi.org/10.1016/j.ceb.2006.06.008
- J. Han, and R.J. Kaufman, "The role of ER stress in lipid metabolism and lipotoxicity", Journal of Lipid Research, vol. 57, pp. 1329-1338, 2016. http://dx.doi.org/10.1194/jlr.R067595
- G. Drin, "Topological Regulation of Lipid Balance in Cells", Annual Review of Biochemistry, vol. 83, pp. 51-77, 2014. http://dx.doi.org/10.1146/annurev-biochem-060713-035307
- S. Goldfarb, "Submicrosomal localization of hepatic 3‐hydroxy‐3‐methylglutaryl coenzyme a (HMG‐CoA) reductase", FEBS Letters, vol. 24, pp. 153-155, 1972. http://dx.doi.org/10.1016/0014-5793(72)80755-5
- E.C. Mandon, I. Ehses, J. Rother, G. van Echten, and K. Sandhoff, "Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver.", The Journal of biological chemistry, 1992. http://www.ncbi.nlm.nih.gov/pubmed/1317856
- G.S. Hotamisligil, "Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease", Cell, vol. 140, pp. 900-917, 2010. http://dx.doi.org/10.1016/j.cell.2010.02.034
- N. Kaplowitz, T.A. Than, M. Shinohara, and C. Ji, "Endoplasmic Reticulum Stress and Liver Injury", Seminars in Liver Disease, vol. 27, pp. 367-377, 2007. http://dx.doi.org/10.1055/s-2007-991513
- H. Malhi, and R.J. Kaufman, "Endoplasmic reticulum stress in liver disease", Journal of Hepatology, vol. 54, pp. 795-809, 2011. http://dx.doi.org/10.1016/j.jhep.2010.11.005
- L.P. Bechmann, R.A. Hannivoort, G. Gerken, G.S. Hotamisligil, M. Trauner, and A. Canbay, "The interaction of hepatic lipid and glucose metabolism in liver diseases", Journal of Hepatology, vol. 56, pp. 952-964, 2012. http://dx.doi.org/10.1016/j.jhep.2011.08.025
- D. Senft, and Z.A. Ronai, "UPR, autophagy, and mitochondria crosstalk underlies the ER stress response", Trends in Biochemical Sciences, vol. 40, pp. 141-148, 2015. http://dx.doi.org/10.1016/j.tibs.2015.01.002
- J.C. Christianson, J.A. Olzmann, T.A. Shaler, M.E. Sowa, E.J. Bennett, C.M. Richter, R.E. Tyler, E.J. Greenblatt, J. Wade Harper, and R.R. Kopito, "Defining human ERAD networks through an integrative mapping strategy", Nature Cell Biology, vol. 14, pp. 93-105, 2011. http://dx.doi.org/10.1038/ncb2383
- J. Stevenson, E.Y. Huang, and J.A. Olzmann, "Endoplasmic Reticulum–Associated Degradation and Lipid Homeostasis", Annual Review of Nutrition, vol. 36, pp. 511-542, 2016. http://dx.doi.org/10.1146/annurev-nutr-071715-051030
- C. Hetz, E. Chevet, and S.A. Oakes, "Proteostasis control by the unfolded protein response", Nature Cell Biology, vol. 17, pp. 829-838, 2015. http://dx.doi.org/10.1038/ncb3184
- D. Lindholm, L. Korhonen, O. Eriksson, and S. Kõks, "Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease", Frontiers in Cell and Developmental Biology, vol. 5, 2017. http://dx.doi.org/10.3389/fcell.2017.00048
- M. Corazzari, M. Gagliardi, G.M. Fimia, and M. Piacentini, "Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate", Frontiers in Oncology, vol. 7, 2017. http://dx.doi.org/10.3389/fonc.2017.00078
- E. Bahar, H. Kim, and H. Yoon, "ER Stress-Mediated Signaling: Action Potential and Ca2+ as Key Players", International Journal of Molecular Sciences, vol. 17, pp. 1558, 2016. http://dx.doi.org/10.3390/ijms17091558
- A. Bertolotti, Y. Zhang, L.M. Hendershot, H.P. Harding, and D. Ron, "Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response", Nature Cell Biology, vol. 2, pp. 326-332, 2000. http://dx.doi.org/10.1038/35014014
- H.P. Harding, Y. Zhang, and D. Ron, "Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase", Nature, vol. 397, pp. 271-274, 1999. http://dx.doi.org/10.1038/16729
- X. Chen, J. Shen, and R. Prywes, "The Luminal Domain of ATF6 Senses Endoplasmic Reticulum (ER) Stress and Causes Translocation of ATF6 from the ER to the Golgi", Journal of Biological Chemistry, vol. 277, pp. 13045-13052, 2002. http://dx.doi.org/10.1074/jbc.M110636200
- J. Shen, E.L. Snapp, J. Lippincott-Schwartz, and R. Prywes, "Stable Binding of ATF6 to BiP in the Endoplasmic Reticulum Stress Response", Molecular and Cellular Biology, vol. 25, pp. 921-932, 2005. http://dx.doi.org/10.1128/MCB.25.3.921-932.2005
- N. Donnelly, A.M. Gorman, S. Gupta, and A. Samali, "The eIF2α kinases: their structures and functions", Cellular and Molecular Life Sciences, vol. 70, pp. 3493-3511, 2013. http://dx.doi.org/10.1007/s00018-012-1252-6
- Y. Ma, J.W. Brewer, J. Alan Diehl, and L.M. Hendershot, "Two Distinct Stress Signaling Pathways Converge Upon the CHOP Promoter During the Mammalian Unfolded Protein Response", Journal of Molecular Biology, vol. 318, pp. 1351-1365, 2002. http://dx.doi.org/10.1016/s0022-2836(02)00234-6
- H.P. Harding, Y. Zhang, D. Scheuner, J. Chen, R.J. Kaufman, and D. Ron, "Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2α) dephosphorylation in mammalian development", Proceedings of the National Academy of Sciences, vol. 106, pp. 1832-1837, 2009. http://dx.doi.org/10.1073/pnas.0809632106
- P.R. Romano, M.T. Garcia-Barrio, X. Zhang, Q. Wang, D.R. Taylor, F. Zhang, C. Herring, M.B. Mathews, J. Qin, and A.G. Hinnebusch, "Autophosphorylation in the Activation Loop Is Required for Full Kinase Activity In Vivo of Human and Yeast Eukaryotic Initiation Factor 2α Kinases PKR and GCN2", Molecular and Cellular Biology, vol. 18, pp. 2282-2297, 1998. http://dx.doi.org/10.1128/mcb.18.4.2282
- W. Tirasophon, K. Lee, B. Callaghan, A. Welihinda, and R.J. Kaufman, "The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response", Genes & Development, vol. 14, pp. 2725-2736, 2000. http://dx.doi.org/10.1101/gad.839400
- H. Yoshida, T. Matsui, A. Yamamoto, T. Okada, and K. Mori, "XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor", Cell, vol. 107, pp. 881-891, 2001. http://dx.doi.org/10.1016/s0092-8674(01)00611-0
- R. Wu, Q. Zhang, Y. Lu, K. Ren, and G. Yi, "Involvement of the IRE1α-XBP1 Pathway and XBP1s-Dependent Transcriptional Reprogramming in Metabolic Diseases", DNA and Cell Biology, vol. 34, pp. 6-18, 2015. http://dx.doi.org/10.1089/dna.2014.2552
- D. Acosta-Alvear, Y. Zhou, A. Blais, M. Tsikitis, N.H. Lents, C. Arias, C.J. Lennon, Y. Kluger, and B.D. Dynlacht, "XBP1 Controls Diverse Cell Type- and Condition-Specific Transcriptional Regulatory Networks", Molecular Cell, vol. 27, pp. 53-66, 2007. http://dx.doi.org/10.1016/j.molcel.2007.06.011
- J. Ye, R.B. Rawson, R. Komuro, X. Chen, U.P. Davé, R. Prywes, M.S. Brown, and J.L. Goldstein, "ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases that Process SREBPs", Molecular Cell, vol. 6, pp. 1355-1364, 2000. http://dx.doi.org/10.1016/s1097-2765(00)00133-7
- C. Hetz, and F.R. Papa, "The Unfolded Protein Response and Cell Fate Control", Molecular Cell, vol. 69, pp. 169-181, 2018. http://dx.doi.org/10.1016/j.molcel.2017.06.017
- S. Song, J. Tan, Y. Miao, M. Li, and Q. Zhang, "Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress", Journal of Cellular Physiology, vol. 232, pp. 2977-2984, 2017. http://dx.doi.org/10.1002/jcp.25785
- Y. Wei, D. Wang, F. Topczewski, and M.J. Pagliassotti, "Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells", American Journal of Physiology-Endocrinology and Metabolism, vol. 291, pp. E275-E281, 2006. http://dx.doi.org/10.1152/ajpendo.00644.2005
- D. Wang, Y. Wei, and M.J. Pagliassotti, "Saturated Fatty Acids Promote Endoplasmic Reticulum Stress and Liver Injury in Rats with Hepatic Steatosis", Endocrinology, vol. 147, pp. 943-951, 2006. http://dx.doi.org/10.1210/en.2005-0570
- P. Puri, F. Mirshahi, O. Cheung, R. Natarajan, J.W. Maher, J.M. Kellum, and A.J. Sanyal, "Activation and Dysregulation of the Unfolded Protein Response in Nonalcoholic Fatty Liver Disease", Gastroenterology, vol. 134, pp. 568-576, 2008. http://dx.doi.org/10.1053/j.gastro.2007.10.039
- M. Mota, B.A. Banini, S.C. Cazanave, and A.J. Sanyal, "Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease", Metabolism, vol. 65, pp. 1049-1061, 2016. http://dx.doi.org/10.1016/j.metabol.2016.02.014
- Y. Liu, M. Shao, Y. Wu, C. Yan, S. Jiang, J. Liu, J. Dai, L. Yang, J. Li, W. Jia, L. Rui, and Y. Liu, "Role for the endoplasmic reticulum stress sensor IRE1α in liver regenerative responses", Journal of Hepatology, vol. 62, pp. 590-598, 2015. http://dx.doi.org/10.1016/j.jhep.2014.10.022
- Y. Inaba, T. Furutani, K. Kimura, H. Watanabe, S. Haga, Y. Kido, M. Matsumoto, Y. Yamamoto, K. Harada, S. Kaneko, S. Oyadomari, M. Ozaki, M. Kasuga, and H. Inoue, "Growth arrest and DNA damage‐inducible 34 regulates liver regeneration in hepatic steatosis in mice", Hepatology, vol. 61, pp. 1343-1356, 2015. http://dx.doi.org/10.1002/hep.27619
- M. Shao, B. Shan, Y. Liu, Y. Deng, C. Yan, Y. Wu, T. Mao, Y. Qiu, Y. Zhou, S. Jiang, W. Jia, J. Li, J. Li, L. Rui, L. Yang, and Y. Liu, "Hepatic IRE1α regulates fasting-induced metabolic adaptive programs through the XBP1s–PPARα axis signalling", Nature Communications, vol. 5, 2014. http://dx.doi.org/10.1038/ncomms4528
- D. Bartoli, D. Piobbico, M.M. Bellet, A.M. Bennati, R. Roberti, M.A. Della Fazia, and G. Servillo, "Impaired cell proliferation in regenerating liver of 3 β-hydroxysterol Δ14-reductase (TM7SF2) knock-out mice", Cell Cycle, vol. 15, pp. 2164-2173, 2016. http://dx.doi.org/10.1080/15384101.2016.1195939
- S. Lee, S. Kim, S. Hwang, N.J. Cherrington, and D. Ryu, "Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease", Oncotarget, vol. 8, pp. 63370-63381, 2017. http://dx.doi.org/10.18632/oncotarget.18812
- C. Ma, Q. Zhang, and T.F. Greten, "Nonalcoholic fatty liver disease promotes hepatocellular carcinoma through direct and indirect effects on hepatocytes", The FEBS Journal, vol. 285, pp. 752-762, 2017. http://dx.doi.org/10.1111/febs.14209
- M.J. Pagliassotti, P.Y. Kim, A.L. Estrada, C.M. Stewart, and C.L. Gentile, "Endoplasmic reticulum stress in obesity and obesity-related disorders: An expanded view", Metabolism, vol. 65, pp. 1238-1246, 2016. http://dx.doi.org/10.1016/j.metabol.2016.05.002
- P.A. Hyslop, D.A. York, and D.L. Corina, "Changes in the composition and fluidity of membranes in obese (ob/ob) mice: a study of hepatic microsomal NADPH-cytochrome P450 oxidoreductase activity.", International journal of obesity, 1982. http://www.ncbi.nlm.nih.gov/pubmed/6811450
- S. Fu, L. Yang, P. Li, O. Hofmann, L. Dicker, W. Hide, X. Lin, S.M. Watkins, A.R. Ivanov, and G.S. Hotamisligil, "Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity", Nature, vol. 473, pp. 528-531, 2011. http://dx.doi.org/10.1038/nature09968
- A. Arruda, and G. Hotamisligil, "Calcium Homeostasis and Organelle Function in the Pathogenesis of Obesity and Diabetes", Cell Metabolism, vol. 22, pp. 381-397, 2015. http://dx.doi.org/10.1016/j.cmet.2015.06.010
- H.L. Kammoun, H. Chabanon, I. Hainault, S. Luquet, C. Magnan, T. Koike, P. Ferré, and F. Foufelle, "GRP78 expression inhibits insulin and ER stress–induced SREBP-1c activation and reduces hepatic steatosis in mice", Journal of Clinical Investigation, vol. 119, pp. 1201-1215, 2009. http://dx.doi.org/10.1172/JCI37007
- J. Lee, R. Mendez, H.H. Heng, Z. Yang, and K. Zhang, "Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation.", American journal of translational research, 2012. http://www.ncbi.nlm.nih.gov/pubmed/22347525
- Y. Wang, J. Viscarra, S. Kim, and H.S. Sul, "Transcriptional regulation of hepatic lipogenesis", Nature Reviews Molecular Cell Biology, vol. 16, pp. 678-689, 2015. http://dx.doi.org/10.1038/nrm4074
- H. Jo, S.S. Choe, K.C. Shin, H. Jang, J.H. Lee, J.K. Seong, S.H. Back, and J.B. Kim, "Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor", Hepatology, vol. 57, pp. 1366-1377, 2013. http://dx.doi.org/10.1002/hep.26126
- G. Alvarez-Sola, I. Uriarte, M.U. Latasa, M.G. Fernandez-Barrena, R. Urtasun, M. Elizalde, M. Barcena-Varela, M. Jiménez, H.C. Chang, R. Barbero, V. Catalán, A. Rodríguez, G. Frühbeck, J.M. Gallego-Escuredo, A. Gavaldà-Navarro, F. Villarroya, C.M. Rodriguez-Ortigosa, F.J. Corrales, J. Prieto, P. Berraondo, C. Berasain, and M.A. Avila, "Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: development of an FGF19-based chimeric molecule to promote fatty liver regeneration", Gut, vol. 66, pp. 1818-1828, 2017. http://dx.doi.org/10.1136/gutjnl-2016-312975
- Y. Yu, M. Tamai, and Y. Tagawa, "Nitric oxide is critical for avoiding hepatic lipid overloading via IL-6 induction during liver regeneration after partial hepatectomy in mice", Experimental Animals, vol. 66, pp. 293-302, 2017. http://dx.doi.org/10.1538/expanim.17-0017
- L. Zhang, F. Ren, X. Zhang, X. Wang, H. Shi, L. Zhou, S. Zheng, Y. Chen, D. Chen, L. Li, C. Zhao, and Z. Duan, "Peroxisome proliferator-activated receptor alpha acts as a mediator of endoplasmic reticulum stress-induced hepatocyte apoptosis in acute liver failure", Disease Models & Mechanisms, 2016. http://dx.doi.org/10.1242/dmm.023242
- C. Enkhbold, Y. Morine, T. Utsunomiya, S. Imura, T. Ikemoto, Y. Arakawa, Y. Saito, S. Yamada, D. Ishikawa, and M. Shimada, "Dysfunction of liver regeneration in aged liver after partial hepatectomy", Journal of Gastroenterology and Hepatology, vol. 30, pp. 1217-1224, 2015. http://dx.doi.org/10.1111/jgh.12930
- I.B. Mosbah, I. Alfany-Fernández, C. Martel, M.A. Zaouali, M. Bintanel-Morcillo, A. Rimola, J. Rodés, C. Brenner, J. Roselló-Catafau, and C. Peralta, "Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia–reperfusion", Cell Death & Disease, vol. 1, pp. e52-e52, 2010. http://dx.doi.org/10.1038/cddis.2010.29
- S. Wada, E. Hatano, T. Yoh, N. Nakamura, Y. Okuda, M. Okuno, Y. Kasai, K. Iwaisako, S. Seo, K. Taura, and S. Uemoto, "CAAT/enhancer binding protein–homologous protein deficiency attenuates liver ischemia/reperfusion injury in mice", Liver Transplantation, vol. 24, pp. 645-654, 2018. http://dx.doi.org/10.1002/lt.25053
- S. Kugel, and R. Mostoslavsky, "Chromatin and beyond: the multitasking roles for SIRT6", Trends in Biochemical Sciences, vol. 39, pp. 72-81, 2014. http://dx.doi.org/10.1016/j.tibs.2013.12.002
- T. Finkel, C. Deng, and R. Mostoslavsky, "Recent progress in the biology and physiology of sirtuins", Nature, vol. 460, pp. 587-591, 2009. http://dx.doi.org/10.1038/nature08197
- E. Michishita, J.Y. Park, J.M. Burneskis, J.C. Barrett, and I. Horikawa, "Evolutionarily Conserved and Nonconserved Cellular Localizations and Functions of Human SIRT Proteins", Molecular Biology of the Cell, vol. 16, pp. 4623-4635, 2005. http://dx.doi.org/10.1091/mbc.E05-01-0033
- F. Nassir, and J.A. Ibdah, "Sirtuins and nonalcoholic fatty liver disease", World Journal of Gastroenterology, vol. 22, pp. 10084, 2016. http://dx.doi.org/10.3748/wjg.v22.i46.10084
- D.B. Lombard, F.W. Alt, H. Cheng, J. Bunkenborg, R.S. Streeper, R. Mostoslavsky, J. Kim, G. Yancopoulos, D. Valenzuela, A. Murphy, Y. Yang, Y. Chen, M.D. Hirschey, R.T. Bronson, M. Haigis, L.P. Guarente, R.V. Farese, S. Weissman, E. Verdin, and B. Schwer, "Mammalian Sir2 Homolog SIRT3 Regulates Global Mitochondrial Lysine Acetylation", Molecular and Cellular Biology, vol. 27, pp. 8807-8814, 2007. http://dx.doi.org/10.1128/MCB.01636-07
- A. Kendrick, M. Choudhury, S. Rahman, C. McCurdy, M. Friederich, J. Van Hove, P. Watson, N. Birdsey, J. Bao, D. Gius, M. Sack, E. Jing, C. Kahn, J. Friedman, and K. Jonscher, "Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation", Biochemical Journal, vol. 433, pp. 505-514, 2011. http://dx.doi.org/10.1042/BJ20100791
- S. Furukawa, T. Fujita, M. Shimabukuro, M. Iwaki, Y. Yamada, Y. Nakajima, O. Nakayama, M. Makishima, M. Matsuda, and I. Shimomura, "Increased oxidative stress in obesity and its impact on metabolic syndrome", Journal of Clinical Investigation, vol. 114, pp. 1752-1761, 2004. http://dx.doi.org/10.1172/JCI21625
- H. Urakawa, A. Katsuki, Y. Sumida, E.C. Gabazza, S. Murashima, K. Morioka, N. Maruyama, N. Kitagawa, T. Tanaka, Y. Hori, K. Nakatani, Y. Yano, and Y. Adachi, "Oxidative Stress Is Associated with Adiposity and Insulin Resistance in Men", The Journal of Clinical Endocrinology & Metabolism, vol. 88, pp. 4673-4676, 2003. http://dx.doi.org/10.1210/jc.2003-030202
- L. Guarente, "Sirtuins, Aging, and Metabolism", Cold Spring Harbor Symposia on Quantitative Biology, vol. 76, pp. 81-90, 2011. http://dx.doi.org/10.1101/sqb.2011.76.010629
- G. Asher, D. Gatfield, M. Stratmann, H. Reinke, C. Dibner, F. Kreppel, R. Mostoslavsky, F.W. Alt, and U. Schibler, "SIRT1 Regulates Circadian Clock Gene Expression through PER2 Deacetylation", Cell, vol. 134, pp. 317-328, 2008. http://dx.doi.org/10.1016/j.cell.2008.06.050
- H. Chang, and L. Guarente, "SIRT1 Mediates Central Circadian Control in the SCN by a Mechanism that Decays with Aging", Cell, vol. 153, pp. 1448-1460, 2013. http://dx.doi.org/10.1016/j.cell.2013.05.027
- Y. Nakahata, M. Kaluzova, B. Grimaldi, S. Sahar, J. Hirayama, D. Chen, L.P. Guarente, and P. Sassone-Corsi, "The NAD+-Dependent Deacetylase SIRT1 Modulates CLOCK-Mediated Chromatin Remodeling and Circadian Control", Cell, vol. 134, pp. 329-340, 2008. http://dx.doi.org/10.1016/j.cell.2008.07.002
- Y. Nakahata, S. Sahar, G. Astarita, M. Kaluzova, and P. Sassone-Corsi, "Circadian Control of the NAD + Salvage Pathway by CLOCK-SIRT1", Science, vol. 324, pp. 654-657, 2009. http://dx.doi.org/10.1126/science.1170803
- M.M. Bellet, Y. Nakahata, M. Boudjelal, E. Watts, D.E. Mossakowska, K.A. Edwards, M. Cervantes, G. Astarita, C. Loh, J.L. Ellis, G.P. Vlasuk, and P. Sassone-Corsi, "Pharmacological modulation of circadian rhythms by synthetic activators of the deacetylase SIRT1", Proceedings of the National Academy of Sciences, vol. 110, pp. 3333-3338, 2013. http://dx.doi.org/10.1073/pnas.1214266110
- S. Masri, P. Rigor, M. Cervantes, N. Ceglia, C. Sebastian, C. Xiao, M. Roqueta-Rivera, C. Deng, T. Osborne, R. Mostoslavsky, P. Baldi, and P. Sassone-Corsi, "Partitioning Circadian Transcription by SIRT6 Leads to Segregated Control of Cellular Metabolism", Cell, vol. 158, pp. 659-672, 2014. http://dx.doi.org/10.1016/j.cell.2014.06.050
- S. Sahar, and P. Sassone-Corsi, "Metabolism and cancer: the circadian clock connection", Nature Reviews Cancer, vol. 9, pp. 886-896, 2009. http://dx.doi.org/10.1038/nrc2747
- R. Ding, J. Bao, and C. Deng, "Emerging roles of SIRT1 in fatty liver diseases", International Journal of Biological Sciences, vol. 13, pp. 852-867, 2017. http://dx.doi.org/10.7150/ijbs.19370
- S.B. Rajamohan, V.B. Pillai, M. Gupta, N.R. Sundaresan, K.G. Birukov, S. Samant, M.O. Hottiger, and M.P. Gupta, "SIRT1 Promotes Cell Survival under Stress by Deacetylation-Dependent Deactivation of Poly(ADP-Ribose) Polymerase 1", Molecular and Cellular Biology, vol. 29, pp. 4116-4129, 2009. http://dx.doi.org/10.1128/MCB.00121-09
- X. Deng, L. Chen, and N. Li, "The expression of SIRT1 in nonalcoholic fatty liver disease induced by high‐fat diet in rats", Liver International, vol. 27, pp. 708-715, 2007. http://dx.doi.org/10.1111/j.1478-3231.2007.01497.x
- A. Purushotham, T.T. Schug, Q. Xu, S. Surapureddi, X. Guo, and X. Li, "Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation", Cell Metabolism, vol. 9, pp. 327-338, 2009. http://dx.doi.org/10.1016/j.cmet.2009.02.006
- T. Ramirez, Y. Li, S. Yin, M. Xu, D. Feng, Z. Zhou, M. Zang, P. Mukhopadhyay, Z.V. Varga, P. Pacher, B. Gao, and H. Wang, "Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression", Journal of Hepatology, vol. 66, pp. 601-609, 2017. http://dx.doi.org/10.1016/j.jhep.2016.11.004
- M.C. Sugden, P.W. Caton, and M.J. Holness, "PPAR control: it's SIRTainly as easy as PGC", Journal of Endocrinology, vol. 204, pp. 93-104, 2009. http://dx.doi.org/10.1677/JOE-09-0359
- B. Ponugoti, D. Kim, Z. Xiao, Z. Smith, J. Miao, M. Zang, S. Wu, C. Chiang, T.D. Veenstra, and J.K. Kemper, "SIRT1 Deacetylates and Inhibits SREBP-1C Activity in Regulation of Hepatic Lipid Metabolism*", Journal of Biological Chemistry, vol. 285, pp. 33959-33970, 2010. http://dx.doi.org/10.1074/jbc.M110.122978
- X. Li, and N. Kazgan, "Mammalian Sirtuins and Energy Metabolism", International Journal of Biological Sciences, vol. 7, pp. 575-587, 2011. http://dx.doi.org/10.7150/ijbs.7.575
- G. Simmons, W. Pruitt, and K. Pruitt, "Diverse Roles of SIRT1 in Cancer Biology and Lipid Metabolism", International Journal of Molecular Sciences, vol. 16, pp. 950-965, 2015. http://dx.doi.org/10.3390/ijms16010950
- J.K. Kemper, S. Choi, and D.H. Kim, "Sirtuin 1 Deacetylase", Vitamins & Hormones, pp. 385-404, 2013. http://dx.doi.org/10.1016/B978-0-12-407766-9.00016-X
- D. Chen, J. Bruno, E. Easlon, S. Lin, H. Cheng, F.W. Alt, and L. Guarente, "Tissue-specific regulation of SIRT1 by calorie restriction", Genes & Development, vol. 22, pp. 1753-1757, 2008. http://dx.doi.org/10.1101/gad.1650608
- R. Wang, C. Li, and C. Deng, "Liver Steatosis and Increased ChREBP Expression in Mice Carrying a Liver Specific SIRT1 Null Mutation under a Normal Feeding Condition", International Journal of Biological Sciences, pp. 682-690, 2010. http://dx.doi.org/10.7150/ijbs.6.682
- S.E. Choi, S. Kwon, S. Seok, Z. Xiao, K. Lee, Y. Kang, X. Li, K. Shinoda, S. Kajimura, B. Kemper, and J.K. Kemper, "Obesity-Linked Phosphorylation of SIRT1 by Casein Kinase 2 Inhibits Its Nuclear Localization and Promotes Fatty Liver", Molecular and Cellular Biology, vol. 37, 2017. http://dx.doi.org/10.1128/MCB.00006-17
- P.T. Pfluger, D. Herranz, S. Velasco-Miguel, M. Serrano, and M.H. Tschöp, "Sirt1 protects against high-fat diet-induced metabolic damage", Proceedings of the National Academy of Sciences, vol. 105, pp. 9793-9798, 2008. http://dx.doi.org/10.1073/pnas.0802917105
- R. Wang, H. Kim, C. Xiao, X. Xu, O. Gavrilova, and C. Deng, "Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance", Journal of Clinical Investigation, vol. 121, pp. 4477-4490, 2011. http://dx.doi.org/10.1172/JCI46243
- M.M. Bellet, S. Masri, G. Astarita, P. Sassone-Corsi, M.A. Della Fazia, and G. Servillo, "Histone Deacetylase SIRT1 Controls Proliferation, Circadian Rhythm, and Lipid Metabolism during Liver Regeneration in Mice", Journal of Biological Chemistry, vol. 291, pp. 23318-23329, 2016. http://dx.doi.org/10.1074/jbc.M116.737114
- S.P. Anderson, L. Yoon, E.B. Richard, C.S. Dunn, R.C. Cattley, and C.J. Corton, "Delayed liver regeneration in peroxisome proliferator-activated receptor-α-null mice", Hepatology, vol. 36, pp. 544-554, 2002. http://dx.doi.org/10.1053/jhep.2002.35276
- X. Zheng, F. Xu, H. Liang, H. Cao, M. Cai, W. Xu, and J. Weng, "SIRT1/HSF1/HSP pathway is essential for exenatide‐alleviated, lipid‐induced hepatic endoplasmic reticulum stress", Hepatology, vol. 66, pp. 809-824, 2017. http://dx.doi.org/10.1002/hep.29238
- Y. Li, S. Xu, A. Giles, K. Nakamura, J.W. Lee, X. Hou, G. Donmez, J. Li, Z. Luo, K. Walsh, L. Guarente, and M. Zang, "Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver", The FASEB Journal, vol. 25, pp. 1664-1679, 2011. http://dx.doi.org/10.1096/fj.10-173492
- S. Ding, J. Jiang, G. Zhang, Y. Bu, G. Zhang, and X. Zhao, "Resveratrol and caloric restriction prevent hepatic steatosis by regulating SIRT1-autophagy pathway and alleviating endoplasmic reticulum stress in high-fat diet-fed rats", PLOS ONE, vol. 12, pp. e0183541, 2017. http://dx.doi.org/10.1371/journal.pone.0183541
- T.W. Jung, K. Lee, M.W. Lee, and K. Ka, "SIRT1 attenuates palmitate-induced endoplasmic reticulum stress and insulin resistance in HepG2 cells via induction of oxygen-regulated protein 150", Biochemical and Biophysical Research Communications, vol. 422, pp. 229-232, 2012. http://dx.doi.org/10.1016/j.bbrc.2012.04.129
- F. Wang, Y. Chen, and H. Ouyang, "Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation", Biochemical Journal, vol. 433, pp. 245-252, 2010. http://dx.doi.org/10.1042/BJ20101293
- T. Koga, M.A. Suico, S. Shimasaki, E. Watanabe, Y. Kai, K. Koyama, K. Omachi, S. Morino-Koga, T. Sato, T. Shuto, K. Mori, S. Hino, M. Nakao, and H. Kai, "Endoplasmic Reticulum (ER) Stress Induces Sirtuin 1 (SIRT1) Expression via the PI3K-Akt-GSK3β Signaling Pathway and Promotes Hepatocellular Injury", Journal of Biological Chemistry, vol. 290, pp. 30366-30374, 2015. http://dx.doi.org/10.1074/jbc.M115.664169
- G. Cretenet, M. Le Clech, and F. Gachon, "Circadian Clock-Coordinated 12 Hr Period Rhythmic Activation of the IRE1α Pathway Controls Lipid Metabolism in Mouse Liver", Cell Metabolism, vol. 11, pp. 47-57, 2010. http://dx.doi.org/10.1016/j.cmet.2009.11.002
- C. Maillo, J. Martín, D. Sebastián, M. Hernández-Alvarez, M. García-Rocha, O. Reina, A. Zorzano, M. Fernandez, and R. Méndez, "Circadian- and UPR-dependent control of CPEB4 mediates a translational response to counteract hepatic steatosis under ER stress", Nature Cell Biology, vol. 19, pp. 94-105, 2017. http://dx.doi.org/10.1038/ncb3461
- J. Soeda, P. Cordero, J. Li, A. Mouralidarane, E. Asilmaz, S. Ray, V. Nguyen, R. Carter, M. Novelli, M. Vinciguerra, L. Poston, P.D. Taylor, and J.A. Oben, "Hepatic rhythmicity of endoplasmic reticulum stress is disrupted in perinatal and adult mice models of high-fat diet-induced obesity", International Journal of Food Sciences and Nutrition, vol. 68, pp. 455-466, 2016. http://dx.doi.org/10.1080/09637486.2016.1261086
- R. Nogueiras, K.M. Habegger, N. Chaudhary, B. Finan, A.S. Banks, M.O. Dietrich, T.L. Horvath, D.A. Sinclair, P.T. Pfluger, and M.H. Tschöp, "Sirtuin 1 and Sirtuin 3: Physiological Modulators of Metabolism", Physiological Reviews, vol. 92, pp. 1479-1514, 2012. http://dx.doi.org/10.1152/physrev.00022.2011
- A. Hebert, K. Dittenhafer-Reed, W. Yu, D. Bailey, E. Selen, M. Boersma, J. Carson, M. Tonelli, A. Balloon, A. Higbee, M. Westphall, D. Pagliarini, T. Prolla, F. Assadi-Porter, S. Roy, J. Denu, and J. Coon, "Calorie Restriction and SIRT3 Trigger Global Reprogramming of the Mitochondrial Protein Acetylome", Molecular Cell, vol. 49, pp. 186-199, 2013. http://dx.doi.org/10.1016/j.molcel.2012.10.024
- M. Hirschey, T. Shimazu, E. Jing, C. Grueter, A. Collins, B. Aouizerat, A. Stančáková, E. Goetzman, M. Lam, B. Schwer, R. Stevens, M. Muehlbauer, S. Kakar, N. Bass, J. Kuusisto, M. Laakso, F. Alt, C. Newgard, R. Farese, C. Kahn, and E. Verdin, "SIRT3 Deficiency and Mitochondrial Protein Hyperacetylation Accelerate the Development of the Metabolic Syndrome", Molecular Cell, vol. 44, pp. 177-190, 2011. http://dx.doi.org/10.1016/j.molcel.2011.07.019
- S. Ka, I.H. Bang, E.J. Bae, and B. Park, "Hepatocyte‐specific sirtuin 6 deletion predisposes to nonalcoholic steatohepatitis by up‐regulation of Bach1, an Nrf2 repressor", The FASEB Journal, vol. 31, pp. 3999-4010, 2017. http://dx.doi.org/10.1096/fj.201700098RR
- E. Ford, R. Voit, G. Liszt, C. Magin, I. Grummt, and L. Guarente, "Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription", Genes & Development, vol. 20, pp. 1075-1080, 2006. http://dx.doi.org/10.1101/gad.1399706
- J. Shin, M. He, Y. Liu, S. Paredes, L. Villanova, K. Brown, X. Qiu, N. Nabavi, M. Mohrin, K. Wojnoonski, P. Li, H. Cheng, A. Murphy, D. Valenzuela, H. Luo, P. Kapahi, R. Krauss, R. Mostoslavsky, G. Yancopoulos, F. Alt, K. Chua, and D. Chen, "SIRT7 Represses Myc Activity to Suppress ER Stress and Prevent Fatty Liver Disease", Cell Reports, vol. 5, pp. 654-665, 2013. http://dx.doi.org/10.1016/j.celrep.2013.10.007
- C. Hetz, E. Chevet, and H.P. Harding, "Targeting the unfolded protein response in disease", Nature Reviews Drug Discovery, vol. 12, pp. 703-719, 2013. http://dx.doi.org/10.1038/nrd3976
ACKNOWLEDGMENTS
We thank Paolo Puccetti, Stefania Pieroni, Danilo Piobbico, Marilena Castelli and Damiano Scopetti for critical reading of the manuscript and helpful discussion. This study was supported by grants from Associazione Umbra Contro il Cancro (AUCC) and Fondazione Cassa di Risparmio di Perugia and Comitato Maria Grazia Frasconi. G.S. is recipient of a PRIN 2015 Project 20152CB22L_004.
COPYRIGHT
© 2018

Foie gras and liver regeneration: a fat dilemma by Della Fazia and Servillo is licensed under a Creative Commons Attribution 4.0 International License.


