News and thoughts:
Cell Stress, Vol. 2, No. 9, pp. 219 - 224; doi: 10.15698/cst2018.09.151
Unweaving the role of nuclear Lamins in neural circuit integrity

1 Program in Developmental Biology, Baylor College of Medicine (BCM), Houston, TX 77030.
2 Department of Molecular and Human Genetics, BCM, Houston, TX 77030.
3 Department of Neuroscience, BCM, Houston, TX 77030.
4 Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX.
Keywords: Lamin, neurodegeneration, dopaminergic neuron, giant fiber circuit, Drosophila.
Received originally: 03/09/2018 Received in revised form: 05/09/2018
Accepted: 05/09/2018
Published: 10/09/2018
Correspondence:
Shinya Yamamoto, Tel: +1-832-824-8119 yamamoto@bcm.edu
Conflict of interest statement: The authors have no conflict of interest to declare.
Please cite this article as: Samantha L. Deal and Shinya Yamamoto (2018). Unweaving the role of nuclear Lamins in neural circuit integrity. Cell Stress 2(9): 219-224. doi: 10.15698/cst2018.09.151
Lamins are type-V intermediate filament proteins that comprise the nuclear lamina. Although once considered static structural components that provide physical support to the inner nuclear envelope, recent studies are revealing additional functional and regulatory roles for Lamins in chromatin organization, gene regulation, DNA repair, cell division and signal transduction. In this issue of Cell Stress, Oyston et al. (2018) reports the function of Lamin in the maintenance of nervous system integrity and neural circuit function using Drosophila. A number of laminopathies in humans exhibit age-dependent neurological phenotypes, but understanding how defects in specific neural cell types or circuitries contribute to patient phenotypes is very challenging. Drosophila provides a simple yet sophisticated system to begin untangling the vulnerability of diverse neuronal cell types and circuits against cellular stressors induced by defects in nuclear lamina organization.
Structural integrity of the nuclear membrane is essential for the homeostasis of eukaryotic cells. Critical components of this process are Lamins or type-V intermediate filament proteins that form a meshwork on the inner nuclear membrane [1]. In fact, mutations in genes that encode Lamins and related proteins cause a class of diseases referred to as ‘laminopathies’. In the human genome, there are three genes (LMNA, LMNB1, LMNB2) that encode four major Lamin proteins, and biochemists have classified these into A-type and B-type based on their structural features and biochemical properties [2]. Two A-type Lamins, Lamin A and Lamin C, are both made from the LMNA gene through alternative splicing, whereas two B-type Lamins, Lamin B1 and Lamin B2, are transcribed from LMNB1 and LMNB2 genes, respectively. One major difference between these two types of Lamins is that type-B Lamins undergo farnesylation at their C-terminal CaaX box that regulates their intranuclear localization, while mature type-A Lamins lack this modification. The two types of Lamins also differ in their expression patterns. While type-B Lamins are ubiquitously expressed throughout development, type-A Lamins are expressed at later stages when cells undergo differentiation [3]. Although most post-differentiated cells express both types of Lamins, some cell types, such as naïve T and B cells of the immune system, lack type-A Lamin expression [4]. Furthermore, type-A and type-B Lamins are not fully redundant; each type of Lamin forms their own intermediate filament networks and have distinct functions in the nucleus [5].
–
Based on OMIM (Online Mendelian Inheritance in Man, https://www.omim.org/) [6], there are 15 types of laminopathies reported in humans that are caused by mutations in the three genes that encode Lamin proteins (Table 1). Mutations in genes that encode proteins that post-translationally modify Lamins (e.g. ZMPSTE24) as well as proteins that physically interact and function with them (e.g. EMD, LBR) can cause other forms of laminopathies. Laminopathies that the LMNA gene are grouped as primary laminopathies, and more than 600 different mutations have been reported to date [7]. Different mutations in LMNA can result in very different phenotypes, including muscular dystrophy, cardiomyopathy, axonal neuropathy, and progeria [8]. Although some patterns have emerged through functional studies of disease-associated mutations (e.g. progeria phenotype is caused by a gain-of-toxic function of aberrant LMNA), a clear-cut genotype-phenotype correlation is yet to be established, adding another layer of complexity in understanding the molecular mechanisms that underlie these disorders. Many laminopathy studies have focused on aging-related symptoms due to the striking premature aging phenotypes observed in Hutchinson-Gilford progeria (MIM #176670) [9]. In addition, many investigators have been interested in the function of Lamins in cardiac and skeletal muscles since most of the laminopathies exhibit striking symptoms in these tissues [10]. In contrast, the function of Lamins in the nervous system, particularly in the aging brain, is still understudied [11]. Conventional as well as conditional gene knockout studies in mice revealed that loss of LmnB1 or LmnB2 led to neurodevelopmental phenotypes [12][13]. Furthermore, double knockout mice for both LmnB1 and LmnB2 exhibited severe brain atrophy and microcephaly [13][14], indicating that type-B Lamins are necessary for neuronal survival. However, it is unclear whether a milder reduction of Lamin levels in the neurons causes neurodegenerative and/or functional consequences. Such experiments would not only enhance our understanding of laminopathies but may also provide insights into more common neurodegenerative diseases such as Alzheimer’s disease, a condition in which reduction of type-B Lamins and subsequent alterations in neuronal nuclear architecture have been implicated in its pathogenesis [15][16].
–
TABLE 1. List of human laminopathies that are caused by mutations in Lamin genes. |
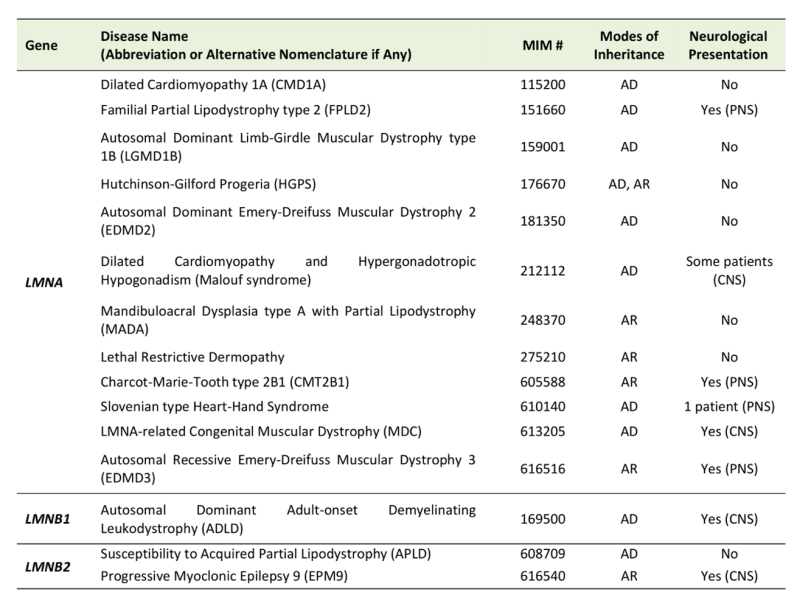 |
Abbreviations: AR (Autosomal Recessive), AD (Autosomal Dominant), CNS (Central Nervous System), PNS (Peripheral Nervous System). Whether the patient of these diseases have been reported to exhibit neurological symptoms have been extracted from OMIM [https://www.omim.org/ (Accessed on 8/31/2018)]. |
–
In addition to vertebrate model systems such as zebrafish, mice and human derived tissues and cells, invertebrate model organisms such as Caenorhabditis elegans [17][18] and Drosophila melanogaster [19][20] have contributed to understanding the in vivo function of Lamins. Based on phylogenetic studies, the last common ancestor of vertebrates and invertebrates (urbilaterian) likely possessed a single Lamin gene that encodes a type-B Lamin protein [21]. C. elegans and most invertebrate species have only one Lamin gene (lmn-1 gene in C. elegans), whereas the D. melanogaster genome carries two genes. Although these genes are thought to have arisen independently from vertebrate Lamin genes, one gene, Lamin C (LamC), shows features of a type-A Lamin whereas the other gene, Lamin (Lam, also referred to as Lamin Dm) has characteristics of a type-B Lamin [22][23]. For example, Lam is expressed ubiquitously and the encoded protein undergoes farnesylation, whereas LamC expression is developmentally regulated and the protein lacks a CaaX motif. Interestingly, LamC is not expressed in the nervous system [24][25], simplifying the functional study of Lamins in the brain of Drosophila.
–
In this issue of Cell Stress, Oyston, Neely and colleagues assessed the functional consequences of reduction of Lamin levels in the nervous system by performing neuronal specific knockdown of Lam in Drosophila [26]. Previous studies in flies have reported shortened lifespan and locomotion defects accompanied by histological signs of neurodegeneration in the brains of hypomorphic Lam mutants [16][25][27]. However, these studies did not examine whether these effects were due to cell-autonomous defects in neurons or due to systemic effects. By combining histological examinations, behavioral assays and electrophysiological measurements in vivo, the authors found that reduction of Lam specifically in post-differentiated neurons causes short lifespan and age-dependent motor deficits, accompanied by loss of subsets of neurons and alterations in specific neural circuit function (Figure 1). Using two independent UAS-RNAi lines against Lam and a neuronal specific GAL4 driver (elavC155-GAL4), which effectively knockdown mRNA and protein levels, Oyston and colleagues identified that flies with reduced neuronal Lam exhibited gradual loss of motor function as measured through a climbing (negative geotaxis) assay. Interestingly, the authors did not observe overt changes in brain morphology that correlated with the motor deficit and shorted lifespan in Lam knockdown flies. Since a previous study reported similar age-dependent motor deterioration upon loss of dopaminergic neurons in the PAM (Protocerebral Anterior Medial) cluster [28], the authors performed immunohistochemical assessment of dopaminergic neurons in the brain. Interestingly, while they did not observe any histological defects in most dopaminergic clusters of the adult brain, they found significant decrease in the number of PAM cluster neurons (Figure 1), consistent with their behavioral data.
–
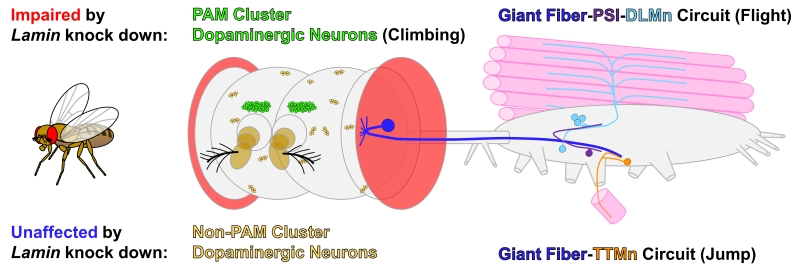 |
FIGURE 1: Specific neurons and neural circuits in Drosophila are more sensitive to reduction of Lamin levels. Schematic diagram of the adult fly central nervous system (brain and thoracicoabdominal ganglion) highlighting the neurons and neural circuits that were studied in Oyston et al. 2018. Eyes, antennae and certain muscles are also depicted as landmarks. For simplicity, only one side of the bilateral giant fiber system is shown here. Flies with decreased neuronal Lamin expression exhibited age-dependent motor deficits and shortened life span. The authors observed histological or functional defects in PAM (protocerebral anterior medial) cluster dopaminergic neurons and in escape response mediating neuronal circuits composed of giant fiber neurons, peripherally synapsing interneurons (PSI) and dorsal longitudinal motor neurons (DLMn). In contrast, non-PAM cluster dopaminergic neurons and a parallel giant fiber circuit comprised of tergo-trochanteral motor neurons (TTMn) were not affected. |
–
To further assess whether the reduction of Lam in the nervous system alters neuron function, the authors turned to the giant fiber system, a well-documented neural circuit that is accessible for in vivo electrophysiological recordings in adult flies [29][30]. The giant fiber system is composed of two parallel circuits that control two groups of muscles that are essential for a rapid escape response (Figure 1). Firing of giant fibers, also known as giant descending neurons, induced through a specific visual stimulus (looming object), triggers simultaneous activation of the two circuits. In the first circuit, giant fibers synapse directly onto motor neurons (TTMn) that regulates the contraction of the tergotrochanteral muscle (TTM), also known as the ‘jump muscle’. The giant fiber-TTMn synapses are primarily mediated by electrical gap junctions (electrical synapse), allowing fast transmission of action potentials. In the second circuit, giant fibers synapse onto peripherally synapsing interneurons (PSI), which in turn synapse onto motor neurons (DLMn) that regulate dorsal longitudinal muscles (DLM), also referred to as the ‘indirect flight muscles’, that are critical for flight. Since the PSI-DLMn connection is predominantly composed of chemical synapses, the response time to trigger the ‘flight program’ is slower compared to the TTMn-mediated ‘jump response’. Hence, the firing of the giant fiber system allows immediate triggering of a quick jump action by the TTM upon identification of an object that is quickly approaching (e.g. a fly swatter), followed by activation of a coordinated flight program through the DLM to fly away from a potential threat. Oyston et al. identified that while Lam reduction led to age-dependent failure in the DLM branch of the giant fiber circuit, interestingly, they did not observe any significant defects in the TTM branch, suggesting that some circuits are more sensitive to reduction of Lam levels than others.
–
The finding that certain neuronal cell types (e.g. PAM cluster dopaminergic neurons) and circuits (e.g. giant fiber-PSI-DLM circuit) in the fly nervous system are more sensitive to the reduction of Lam provide a novel basis for understanding the role of Lamins in the nervous system. It is possible that the differences in the vulnerability may be arising due to the differences in endogenous expression levels of Lam in different cell types or through differences in the knockdown efficiency of Lam via RNAi (e.g. difference in the levels of GAL4 expression, efficiency of dsRNA processing), which needs to be further tested using different GAL4 drivers and other genetic tools. However, it is interesting to hypothesize that Lam may play different roles in different neuronal cell types to maintain the cells’ function and health. In mice, the complete loss of type-B lamins in cone photoreceptors (HRGP-Cre Lmnb1Δ/ΔLmnb2Δ/Δ) do not cause any major morphological or functional defects [31], while a similar manipulation in the forebrain (Emx1-Cre Lmnb1Δ/ΔLmnb2Δ/Δ) leads to atrophy of the cortex and hippocampus [13]. Hence, the concept of different neuronal populations having different vulnerabilities to reduction of Lamin levels may be evolutionarily conserved. In their manuscript, Oyston et al. propose that neural circuits that depend on chemical synapses may be more sensitive to Lamin loss based on the difference they observed between DLM and TTM. While this is an interesting theory, additional electrophysiological studies in other neural circuits that require electrical and chemical synapses will be critical to determining whether this observation can be generalized beyond the giant fiber system. In the manuscript, the authors also propose that the defect they observe in the DLM pathway may be due to loss of neuromuscular junction integrity, or through trans-synaptic effects that alter the responsiveness of the post-synaptic muscle. Future studies investigating the histological and ultrastructural features that are caused by the reduction of Lam in the giant fiber system will likely clarify the mechanism that underlies the age-dependent functional deterioration of the DLM branch of the giant fiber system. In the Xenopus visual system, mRNA for Lamin B2 is transported to axons of retinal ganglion cells for local translation [32]. In this context, Lamin B2 is translated in distal axons and localizes to the mitochondria where it supports axonal integrity by maintaining proper mitochondrial membrane potential and morphology, as well as axonal transport of lysosomes. Further investigation of the Drosophila giant fiber system focusing on the potential extra-nuclear function of Lam in axonal integrity may provide additional molecular insights into the role of Lamins in neural circuit maintenance.
–
In summary, the data presented by Oyston et al. revealed that neuronal specific reduction of Lam recapitulates the neurodegenerative phenotype seen in hypomorphic Lam mutants. Their finding that some cells and neural circuits are more vulnerable to the reduction of Lam levels provides important insights into our understanding of age-dependent neuronal phenotypes observed in a subset of laminopathies in humans. For example, two individuals from a consanguineous family that presented with progressive myoclonic epilepsy 9 (MIM #616540) were found to be homozygous for a functional missense mutation (p.H157Y) in LMNB2 [33]. Epilepsy in these patients began when they were 6 or 7 years old, respectively, and progressed over time. While these phenotypes may be due to developmental defects in neuronal migration as reported in Lmnb2 knockout mice, differences in vulnerability for LMNB2 loss-of-function in post-differentiated neurons may also contribute to phenotypes that appear and progress in an age-dependent manner. In addition, duplications [34] and regulatory mutations that increase the expression of LMNB1 [35] cause adult-onset autosomal dominant leukodystrophy (MIM #169500), a slow progressive neurodegenerative disease. Future complementary studies in Drosophila could assess whether certain cells and circuits are more vulnerable to increases in the level of type-B Lamins and thus may provide insights into why certain neuronal functions are affected more than others in this disorder.
References
- R. de Leeuw, Y. Gruenbaum, and O. Medalia, "Nuclear Lamins: Thin Filaments with Major Functions", Trends in Cell Biology, vol. 28, pp. 34-45, 2018. http://dx.doi.org/10.1016/j.tcb.2017.08.004
- S.A. Adam, and R.D. Goldman, "Insights into the differences between the A- and B-type nuclear lamins", Advances in Biological Regulation, vol. 52, pp. 108-113, 2012. http://dx.doi.org/10.1016/j.advenzreg.2011.11.001
- R.A. Röber, K. Weber, and M. Osborn, "Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study.", Development (Cambridge, England), 1989. http://www.ncbi.nlm.nih.gov/pubmed/2680424
- R.A. Röber, H. Sauter, K. Weber, and M. Osborn, "Cells of the cellular immune and hemopoietic system of the mouse lack lamins A/C: distinction versus other somatic cells.", Journal of cell science, 1990. http://www.ncbi.nlm.nih.gov/pubmed/2200797
- Y. Gruenbaum, and R. Foisner, "Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation", Annual Review of Biochemistry, vol. 84, pp. 131-164, 2015. http://dx.doi.org/10.1146/annurev-biochem-060614-034115
- J.S. Amberger, C.A. Bocchini, F. Schiettecatte, A.F. Scott, and A. Hamosh, "OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders", Nucleic Acids Research, vol. 43, pp. D789-D798, 2014. http://dx.doi.org/10.1093/nar/gku1205
- M. Tatli, and O. Medalia, "Insight into the functional organization of nuclear lamins in health and disease", Current Opinion in Cell Biology, vol. 54, pp. 72-79, 2018. http://dx.doi.org/10.1016/j.ceb.2018.05.001
- S. Kang, M. Yoon, and B. Park, "Laminopathies; Mutations on single gene and various human genetic diseases", BMB Reports, vol. 51, pp. 327-337, 2018. http://dx.doi.org/10.5483/bmbrep.2018.51.7.113
- F.L. DeBusk, "The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature.", The Journal of pediatrics, 1972. http://www.ncbi.nlm.nih.gov/pubmed/4552697
- F. Azibani, A. Muchir, N. Vignier, G. Bonne, and A.T. Bertrand, "Striated muscle laminopathies", Seminars in Cell & Developmental Biology, vol. 29, pp. 107-115, 2014. http://dx.doi.org/10.1016/j.semcdb.2014.01.001
- S.G. Young, H. Jung, J.M. Lee, and L.G. Fong, "Nuclear Lamins and Neurobiology", Molecular and Cellular Biology, vol. 34, pp. 2776-2785, 2014. http://dx.doi.org/10.1128/MCB.00486-14
- C. Coffinier, S.Y. Chang, C. Nobumori, Y. Tu, E.A. Farber, J.I. Toth, L.G. Fong, and S.G. Young, "Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency", Proceedings of the National Academy of Sciences, vol. 107, pp. 5076-5081, 2010. http://dx.doi.org/10.1073/pnas.0908790107
- C. Coffinier, H. Jung, C. Nobumori, S. Chang, Y. Tu, R.H. Barnes, Y. Yoshinaga, P.J. de Jong, L. Vergnes, K. Reue, L.G. Fong, and S.G. Young, "Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons", Molecular Biology of the Cell, vol. 22, pp. 4683-4693, 2011. http://dx.doi.org/10.1091/mbc.E11-06-0504
- Y. Kim, A.A. Sharov, K. McDole, M. Cheng, H. Hao, C. Fan, N. Gaiano, M.S.H. Ko, and Y. Zheng, "Mouse B-Type Lamins Are Required for Proper Organogenesis But Not by Embryonic Stem Cells", Science, vol. 334, pp. 1706-1710, 2011. http://dx.doi.org/10.1126/science.1211222
- B. Frost, "Alzheimer's disease: An acquired neurodegenerative laminopathy", Nucleus, vol. 7, pp. 275-283, 2016. http://dx.doi.org/10.1080/19491034.2016.1183859
- B. Frost, F. Bardai, and M. Feany, "Lamin Dysfunction Mediates Neurodegeneration in Tauopathies", Current Biology, vol. 26, pp. 129-136, 2016. http://dx.doi.org/10.1016/j.cub.2015.11.039
- E.M. Bank, K. Ben-Harush, N. Wiesel-Motiuk, R. Barkan, N. Feinstein, O. Lotan, O. Medalia, and Y. Gruenbaum, "A laminopathic mutation disrupting lamin filament assembly causes disease-like phenotypes inCaenorhabditis elegans", Molecular Biology of the Cell, vol. 22, pp. 2716-2728, 2011. http://dx.doi.org/10.1091/mbc.E11-01-0064
- A. Mattout, B. Pike, B. Towbin, E. Bank, A. Gonzalez-Sandoval, M. Stadler, P. Meister, Y. Gruenbaum, and S. Gasser, "An EDMD Mutation in C. elegans Lamin Blocks Muscle-Specific Gene Relocation and Compromises Muscle Integrity", Current Biology, vol. 21, pp. 1603-1614, 2011. http://dx.doi.org/10.1016/j.cub.2011.08.030
- H. Chen, X. Zheng, and Y. Zheng, "Age-Associated Loss of Lamin-B Leads to Systemic Inflammation and Gut Hyperplasia", Cell, vol. 159, pp. 829-843, 2014. http://dx.doi.org/10.1016/j.cell.2014.10.028
- R. Uchino, Y. Nonaka, T. Horigome, S. Sugiyama, and K. Furukawa, "Loss of Drosophila A-type lamin C initially causes tendon abnormality including disintegration of cytoskeleton and nuclear lamina in muscular defects", Developmental Biology, vol. 373, pp. 216-227, 2013. http://dx.doi.org/10.1016/j.ydbio.2012.08.001
- A. Peter, and R. Stick, "Evolution of the lamin protein family", Nucleus, vol. 3, pp. 44-59, 2012. http://dx.doi.org/10.4161/nucl.18927
- M. Pałka, A. Tomczak, K. Grabowska, M. Machowska, K. Piekarowicz, D. Rzepecka, and R. Rzepecki, "Laminopathies: what can humans learn from fruit flies", Cellular & Molecular Biology Letters, vol. 23, 2018. http://dx.doi.org/10.1186/s11658-018-0093-1
- S.R. Schulze, B. Curio-Penny, S. Speese, G. Dialynas, D.E. Cryderman, C.W. McDonough, D. Nalbant, M. Petersen, V. Budnik, P.K. Geyer, and L.L. Wallrath, "A Comparative Study of Drosophila and Human A-Type Lamins", PLoS ONE, vol. 4, pp. e7564, 2009. http://dx.doi.org/10.1371/journal.pone.0007564
- B. Lenz-Böhme, J. Wismar, S. Fuchs, R. Reifegerste, E. Buchner, H. Betz, and B. Schmitt, "Insertional Mutation of the Drosophila Nuclear Lamin Dm0 Gene Results in Defective Nuclear Envelopes, Clustering of Nuclear Pore Complexes, and Accumulation of Annulate Lamellae", The Journal of Cell Biology, vol. 137, pp. 1001-1016, 1997. http://dx.doi.org/10.1083/jcb.137.5.1001
- S. Osouda, Y. Nakamura, B. de Saint Phalle, M. McConnell, T. Horigome, S. Sugiyama, P.A. Fisher, and K. Furukawa, "Null mutants of Drosophila B-type lamin Dm show aberrant tissue differentiation rather than obvious nuclear shape distortion or specific defects during cell proliferation", Developmental Biology, vol. 284, pp. 219-232, 2005. http://dx.doi.org/10.1016/j.ydbio.2005.05.022
- L.J. Oyston, Y.Q. Lin, T.M. Khuong, Q. Wang, M.T. Lau, T. Clark, and G.G. Neely, "Neuronal Lamin regulates motor circuit integrity and controls motor function and lifespan", Cell Stress, vol. 2, pp. 225-232, 2018. http://dx.doi.org/10.15698/cst2018.09.152
- A. Muñoz-Alarcón, M. Pavlovic, J. Wismar, B. Schmitt, M. Eriksson, P. Kylsten, and M.S. Dushay, "Characterization of lamin Mutation Phenotypes in Drosophila and Comparison to Human Laminopathies", PLoS ONE, vol. 2, pp. e532, 2007. http://dx.doi.org/10.1371/journal.pone.0000532
- T. Riemensperger, A. Issa, U. Pech, H. Coulom, M. Nguyễn, M. Cassar, M. Jacquet, A. Fiala, and S. Birman, "A Single Dopamine Pathway Underlies Progressive Locomotor Deficits in a Drosophila Model of Parkinson Disease", Cell Reports, vol. 5, pp. 952-960, 2013. http://dx.doi.org/10.1016/j.celrep.2013.10.032
- M.J. Allen, and T.A. Godenschwege, "Electrophysiological Recordings from the Drosophila Giant Fiber System (GFS)", Cold Spring Harbor Protocols, vol. 2010, pp. pdb.prot5453, 2010. http://dx.doi.org/10.1101/pdb.prot5453
- V. Martinez, C. Javadi, E. Ngo, L. Ngo, R. Lagow, and B. Zhang, "Age‐related changes in climbing behavior and neural circuit physiology in Drosophila", Developmental Neurobiology, vol. 67, pp. 778-791, 2007. http://dx.doi.org/10.1002/dneu.20388
- D. Razafsky, C. Ward, C. Potter, W. Zhu, Y. Xue, V.J. Kefalov, L.G. Fong, S.G. Young, and D. Hodzic, "Lamin B1 and lamin B2 are long-lived proteins with distinct functions in retinal development", Molecular Biology of the Cell, vol. 27, pp. 1928-1937, 2016. http://dx.doi.org/10.1091/mbc.E16-03-0143
- B. Yoon, H. Jung, A. Dwivedy, C. O'Hare, K. Zivraj, and C. Holt, "Local Translation of Extranuclear Lamin B Promotes Axon Maintenance", Cell, vol. 148, pp. 752-764, 2012. http://dx.doi.org/10.1016/j.cell.2011.11.064
- J.A. Damiano, Z. Afawi, M. Bahlo, M. Mauermann, A. Misk, T. Arsov, K.L. Oliver, H.M. Dahl, A.E. Shearer, R.J. Smith, N.E. Hall, K. Mahmood, R.J. Leventer, I.E. Scheffer, M. Muona, A. Lehesjoki, A.D. Korczyn, H. Herrmann, S.F. Berkovic, and M.S. Hildebrand, "Mutation of the nuclear lamin geneLMNB2in progressive myoclonus epilepsy with early ataxia", Human Molecular Genetics, vol. 24, pp. 4483-4490, 2015. http://dx.doi.org/10.1093/hmg/ddv171
- Q.S. Padiath, K. Saigoh, R. Schiffmann, H. Asahara, T. Yamada, A. Koeppen, K. Hogan, L.J. Ptáček, and Y. Fu, "Lamin B1 duplications cause autosomal dominant leukodystrophy", Nature Genetics, vol. 38, pp. 1114-1123, 2006. http://dx.doi.org/10.1038/ng1872
- E. Giorgio, D. Robyr, M. Spielmann, E. Ferrero, E. Di Gregorio, D. Imperiale, G. Vaula, G. Stamoulis, F. Santoni, C. Atzori, L. Gasparini, D. Ferrera, C. Canale, M. Guipponi, L.A. Pennacchio, S.E. Antonarakis, A. Brussino, and A. Brusco, "A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD)", Human Molecular Genetics, vol. 24, pp. 3143-3154, 2015. http://dx.doi.org/10.1093/hmg/ddv065
ACKNOWLEDGMENTS
We thank Dr. Hillary K. Graves for critical reading of this manuscript. SY has been support-ed by research funds from the NIH (Grant# U54NS093793), Simons Foundation Autism Research Initiative (Grant# 368479), Baylor College of Medicine (Naman Family Fund for Basic Research and Caroline Wiess Law Fund for Research in Molecular Medicine), and Texas Children’s Hospital (Jan and Dan Duncan Neu-rological Research Institute Fellowship).
COPYRIGHT
© 2018

Unweaving the role of nuclear Lamins in neural circuit integrity by Deal and Yamamoto is licensed under a Creative Commons Attribution 4.0 International License.


