Research Articles:
Cell Stress, Vol. 1, No. 1, pp. 55 - 67; doi: 10.15698/cst2017.10.106
mIDH-associated DNA hypermethylation in acute myeloid leukemia reflects differentiation blockage rather than inhibition of TET-mediated demethylation
1 Division of Epigenetics, DKFZ-ZMBH Alliance, German Cancer Research Center; 69120 Heidelberg, Germany.
2 German Consortium of Translational Cancer Research (DKTK), Clinical Cooperation Unit Neuropathology, German Cancer Research Center (DKFZ), Heidelberg, Germany.
3 Department of Neuropathology, Institute of Pathology, Ruprecht-Karls-University Heidelberg, Heidelberg, Germany.
Keywords: mIDH, DNA methylation, AML, TET enzymes, differentiation, D-2-hydroxyglutarate.
Abbreviations:
2-HG – 2-hydroxyglutarate,
5hmC – 5-hydroxymethylcytosine,
AML – acute myeloid leukemia,
CIMP – CpG island methylator phenotype,
DMP – differentially methylated probe,
FAB – French-American-British,
IDH – isocitrate dehydrogenase,
LAD – lamina-associated domain,
mIDH – mutant IDH,
PCA – Principal Component Analysis.
Received originally: 01/08/2017 Received in revised form: 01/09/2017
Accepted: 08/09/2017
Published: 20/09/2017
Correspondence:
Frank Lyko, Deutsches Krebsforschungszentrum (DKFZ), Im Neuenheimer Feld 580, 69120 Heidelberg, Germany; phone: +49-6221-423800; fax: +49-6221-423802 f.lyko@dkfz.de
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Laura Wiehle, Günter Raddatz, Stefan Pusch, Julian Gutekunst, Andreas von Deimling, Manuel Rodríguez-Paredes and Frank Lyko (2017). mIDH-associated DNA hypermethylation in acute myeloid leukemia reflects differentiation blockage rather than inhibition of TET-mediated demethylation. Cell Stress 1(1): 55-67. doi: 10.15698/cst2017.10.106
Abstract
Isocitrate dehydrogenases 1 and 2 (IDH1/2) are recurrently mutated in acute myeloid leukemia (AML), but their mechanistic role in leukemogenesis is poorly understood. The inhibition of TET enzymes by D-2-hydroxyglutarate (D-2-HG), which is produced by mutant IDH1/2 (mIDH1/2), has been suggested to promote epigenetic deregulation during tumorigenesis. In addition, mIDH also induces a differentiation block in various cell culture and mouse models. Here we analyze the genomic methylation patterns of AML patients with mIDH using Infinium 450K data from a large AML cohort and found that mIDH is associated with pronounced DNA hypermethylation at tens of thousands of CpGs. Interestingly, however, myeloid leukemia cells overexpressing mIDH, cells that were cultured in the presence of D-2-HG or TET2 mutant AML patients did not show similar methylation changes. In further analyses, we also characterized the methylation landscapes of myeloid progenitor cells and analyzed their relationship to mIDH-associated hypermethylation. Our findings identify the differentiation state of myeloid cells, rather than inhibition of TET-mediated DNA demethylation, as a major factor of mIDH-associated hypermethylation in AML. Furthermore, our results are also important for understanding the mode of action of currently developed mIDH inhibitors.
INTRODUCTION
Isocitrate dehydrogenase enzymes IDH1 and IDH2 are two closely related enzymes that convert isocitrate to α-ketoglutarate in the tricarboxylic acid (TCA) cycle. The genes encoding IDH1 and IDH2 are recurrently mutated in different cancer entities, such as tumors of the central nervous system, acute myeloid leukemia (AML), cholangiocarcinoma and others [1]. Cancer-associated mutations occur at specific amino acids of the active site and confer a neomorphic function to IDH, resulting in the production of D-2-hydroxyglutarate (D-2-HG;[2]). This metabolite has been suggested to competitively inhibit α-ketoglutarate dependent dioxygenases, including protein hydroxylases, histone demethylases and DNA demethylases [3][4]. Furthermore, mIDH enzymes lose the ability to produce α-ketoglutarate and NADPH [5], resulting in the disturbance of the TCA cycle and cellular metabolic homeostasis. Consequently, defects of various cellular processes such as differentiation, epigenetic modification, growth factor dependence, DNA damage response, hypoxia signaling, mitochondrial respiration, and apoptosis have been reported in the presence of mIDH proteins [5][6][7][8][9][10][11].
–
Abnormal DNA methylation patterns have been described as hallmarks of many cancers [12]. In human AML and lower-grade glioma, global pathogenic DNA hypermethylation has been associated with the presence of mIDH [10][13][14][15][16][17][18][19][20]. The “CpG island methylator phenotype” (CIMP), a specific hypermethylation signature of CpG islands observed in a subset of cancer entities, was suggested to be directly caused by mIDH1 expression in glioma [21]. Mechanistically, it was hypothesized that TET enzymes, which rely on α-ketoglutarate to initiate DNA demethylation via oxidative conversion of 5-methylcytosine to 5-hydroxymethylcytosine (5hmC), might be inhibited by D-2-HG-producing mIDH and thus be responsible for the observed genomic hypermethylation [15][21]. In this context, a critical tumor promoting role was attributed to mIDH-dependent DNA hypermethylation.
–
Mutated IDH enzymes have also become attractive candidates for oncology drug development and several small molecule inhibitors are currently tested in preclinical and clinical research [17][22][23][24][25][26][27][28][29][30][31]. In this context, the reversal of mIDH-associated phenotypes and methylation patterns is considered a key molecular endpoint and biomarker [9][17][32]. However, the reported effects of mIDH inhibitors on genomic DNA methylation patterns appeared to be rather moderate [17][22][23][26]. These observations suggest that mIDH enzymes might promote tumor growth through mechanisms other than the reported inhibition of TET enzymes [33]. Furthermore, these findings emphasize the need to better understand the contribution of mIDH to DNA methylation changes and ultimately tumorigenesis.
–
DNA methylation is a dynamic epigenetic modification that undergoes widespread changes during mammalian development, cellular lineage commitment and differentiation [34]. The hematopoietic system is an excellent model for the analysis of differentiation-associated changes of methylation patterns due to its well characterized differentiation stages and corresponding surface markers allowing the examination of homogeneous primary cell populations [35][36]. The methylome analysis of sorted human hematopoietic cells has demonstrated directional changes with a global loss of methylation during myeloid differentiation [37]. Hypomethylation during myelopoiesis mainly affects differentiation genes with specific functions in the mature cell types as well as transcription factor binding sites and lineage-specific enhancers [37][38][39]. In AML, cells of the stem cell or myeloid progenitor cell pool are malignantly transformed and proliferate aberrantly [40].
–
Previous analyses of mIDH-dependent DNA methylation changes in AML compared unsorted bone marrow aspirates to healthy donor tissue and partially relied on low CpG coverage assays [13][15]. To investigate direct mIDH-, mutant TET- and D-2-HG-dependent DNA methylation changes we analyzed methylation array data of AML patients and HL-60 cells expressing mIDH. The presence of mIDH in AML patients was associated with genomic hypermethylation. However, this effect could not be reproduced in IDH2 R140Q expressing or D-2-HG treated HL-60 cells or in TET2 mutant patients. Instead, we found that methylation landscapes of mIDH carrying patients resembled normal myeloid progenitor cell methylomes. This suggests that altered differentiation states rather than inhibition of DNA demethylation define the mIDH-dependent methylation landscape.
RESULTS
Genomic hypermethylation in AML patients with mIDH
To characterize the methylation patterns associated with neomorphic IDH mutations, we analyzed a published dataset of adult de novo AML [20]. These patients had been clinically annotated and their bone marrow aspirates had been subjected to whole-genome or whole-exome sequencing, RNA sequencing and DNA methylation analysis using Illumina Infinium HumanMethylation450 arrays. We extracted methylation profiles of 28 patients with mutations in either IDH1 (R132) or IDH2 (R140/R172) – herein referred to as mIDH – and 112 patients with IDH wildtype (WT) status. The raw intensity data files were normalized, quality-filtered and statistically analyzed using a standard analytical pipeline [41].
–
Comparison of the two patient groups by unsupervised Principal Component Analysis (PCA) using all 450K probes separated mIDH from IDH WT patients with few exceptions (Fig. 1A), suggesting the presence of specific changes in mIDH-associated methylation profiles. We also performed hierarchical clustering based on the 5000 most significantly differentially methylated (adj. P<0.05) probes. This again correctly segregated the majority of the AML patients according to IDH mutational status, with prominent hypermethylation in the cluster defined by mIDH patients (Fig. 1B). To assess the methylation changes in mIDH patients in more detail, we plotted the beta values of all 70,137 significantly differentially methylated (adj. P<0.05) probes in the two patient groups. The vast majority (68,863) of these probes appeared hypermethylated in patients with mIDH (Fig. 1C). Most differentially methylated probes had an intermediate methylation level in IDH WT AML patients which was increased by approximately 20% in mIDH patients and this increase was highly significant (Fig. 1D; P<0.0001). Our analysis of epigenomic features in mIDH and IDH WT patients revealed that CpG islands, shores, shelves and open sea were all significantly hypermethylated in mIDH patients (Fig. 1E).
–
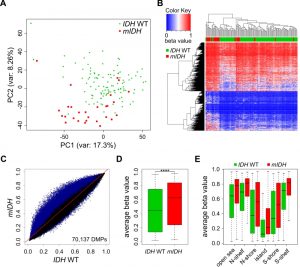 | FIGURE 1: AML patients with mIDH display genomic hypermethylation. (A) Principal Component Analysis of methylomes of 28 AML patients with mIDH and 112 AML patients with IDH WT status using all 450K probes retained after quality filtering. (B) Heatmap of the 5000 most significantly (adj. P<0.05) differentially methylated 450K probes between the two patient groups. Each column represents one patient and each row one probe. Dendrograms of patients and probes were obtained using hierarchical clustering by similarity. The color scale indicates beta values. (C) Scatterplot of the beta values of all 450K probes comparing mIDH to IDH WT patients. Each probe retained after quality filtering is represented by a dot, with significantly (adj. P<0.05) differentially methylated probes (DMPs) depicted in blue. (D) Boxplot showing the average beta values of all the significantly differentially methylated probes in the two groups. The difference between the two groups was highly significant (**** P<0.0001). (E) Boxplot showing the average beta values of the significantly differentially methylated probes associated with different epigenomic features in mIDH and IDH WT patients. All differences observed between the two patient groups were highly significant (P<0.0001). |
–
We also analyzed the possibility that mutations in DNMT3A might influence our PCA and clustering analysis. We therefore compared 32 DNMT3A mutated patients (12 had co-occurring mIDH) to 108 patients with wildtype DNMT3A (20 had co-occurring mIDH) from this cohort. The general effect of DNMT3A mutation was genomic hypo-methylation (23,795 hypomethylated out of 26,334 differentially methylated probes), leading to a modest, but significant reduction of average beta values (Figure S1A, B).
–
Interestingly, many of the outliers in our original PCA (Fig. 1A) had co-occurring DNMT3A mutations (Figure S1C) and when mDNMT3A patients were removed from the initial patient set for hierarchical clustering, segregation according to IDH mutation status was further improved (Figure S1D). Taken together, these results confirm widespread CpG hypermethylation associated with neomorphic IDH1/2 mutations in AML.
–
Methylation analysis of a cellular model expressing mIDH2
In parallel experiments, we generated HL-60 leukemia cells that had either mIDH2 (R140Q) or the corresponding empty vector stably integrated into their genomes. Introduction of mIDH2 resulted in a fivefold increase of IDH2 mRNA expression compared to empty vector (Figure S2A). Consistently, D-2-HG levels in the cell culture medium of mIDH2 expressing cells were elevated more than 30-fold to approximately 11 µM relative to cells transduced with empty vector (Figure S2B). Interestingly, this did not lead to a reduction of TET-dependent global 5hmC levels (Fig. S2C). Furthermore, mIDH2 expressing HL-60 cells showed no overt morphological changes (Figure S3A) and cumulative population doubling measurements indicated that mIDH2 expressing cells had an unchanged proliferation rate (Figure S3B). However, expression analysis of hematopoietic stem cell and myeloid differentiation genes indicated several notable changes: Markers of differentiated myeloid cell states such as CD11B were downregulated, while genes expressed in myeloid progenitor cells such as C-KIT were upregulated in mIDH2 expressing cells compared to the empty vector (Fig. 2A), indicating that mIDH2 expressing HL-60 cells display gene expression changes reminiscent of a less differentiated state. Infinium methylation analysis of two biological replicates per condition showed diverse methylation changes with both hyper- and hypomethylation in HL-60 cells expressing mIDH2 (Fig. 2B), that were clearly different from the specific hypermethylation observed in mIDH AML patients (compare to Fig. 1C).
–
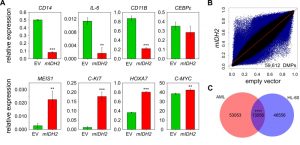 | FIGURE 2: Genomic methylation profiles of mIDH AML patients and HL-60 cells expressing IDH2 R140Q are distinct. (A) Expression analysis of myeloid progenitor and differentiation genes by qRT-PCR in HL-60 cells transduced with empty vector (EV) or mIDH2. Expression is shown relative to ACTB transcript levels. Error bars indicate standard deviation (n=3). Statistical significance was calculated using a two-sided Student's t-test (*** P<0.001, ** P<0.01). (B) Scatterplots comparing average beta values for all probes common to 450K and EPIC chip between HL-60 cells transduced with mIDH2 or empty vector. Each dot represents one probe with DMPs colored in blue. Two biological replicates were analyzed independently on the 450K chip per condition. (C) Venn diagram showing the overlap between DMPs found upon presence of mIDH in the AML patient dataset and the HL-60 cells. The overlap between the two groups was minor, but greater than expected by chance using the hypergeometric test (**** P<0.0001). |
–
Partially methylated domains are known to acquire hypomethylation in cultured cells [42] and coincide with lamina-associated domains (LADs; [43]). To exclude that these regions are responsible for the observed hypomethylation in mIDH2 cells, we removed LAD-associated probes from our analysis. This did not affect the overall result (Fig. S4), indicating that hypomethylation upon expression of mIDH2 occurs outside of LADs. We also identified the probes that were commonly differentially methylated in mIDH AML patients and mIDH2 HL-60 cells, which failed to identify a major overlap (Fig. 2C). Together, these results suggest that the specific DNA hypermethylation pattern observed in mIDH patients cannot be faithfully recapitulated by mIDH2 overexpression in a cell-based model.
–
TET2 mutations in AML are not associated with global DNA hypermethylation
It has been suggested that TET-mediated DNA demethylation is inhibited by mutations in IDH genes, but the observed (hydroxy)methylation changes were determined by indirect assays and appeared rather moderate [15][21]. In our hands, overexpression of mIDH2 in HL-60 cells also did not influence 5hmC abundance in genomic DNA (Fig. S2C), suggesting an oxidation-independent mechanism of mIDH-associated hypermethylation. Additional studies have found contradictory results with regard to methylation changes in TET2 mutated myeloid malignancies [44][45][46]. To analyze potential TET-dependent hypermethylation effects, we extracted patients with mutations in the TET2 gene from the TCGA cohort. These 12 patients presented with nonsense or frameshift mutations occurring in front of the C-terminal catalytic domain, thus destroying the enzymatic function of TET2. The comparison of methylation patterns from the 12 TET2 mutant patients to 100 TET2 and IDH1/2 WT patients did not separate TET2 WT and TET2 mutant patients into distinct clusters in a PCA with all probes (Fig. 3A). Also, we identified only a small fraction of differentially methylated probes (Fig. 3B) which resulted in a very minor and statistically not significant (P=0.52) increase in the average methylation ratio of TET2 mutant patients (Fig. 3C). Together, these data suggest that there are no systematic large-scale DNA methylation changes associated with TET2 mutations in AML, which contrasts the observed mIDH-associated differences. Consequently, it appears unlikely that inhibition of TET2 is a major factor contributing to mIDH-associated DNA hypermethylation.
–
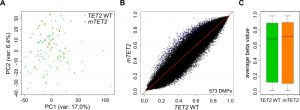 | FIGURE 3: mIDH-associated hypermethylation is not replicated by TET2 mutation in AML patients. (A) Principal Component Analysis of TET2 WT and TET2 mu-tant AML patients using all probes retained after quality filtering. No clear separation of the two patient groups was achieved. (B) Comparison of AML patients with and without TET2 mutations by scatterplot. The average beta value of each 450K probe in the two groups is represented as a single dot. DMPs are colored in blue. IDH mutant patients were removed from the analysis. (C) Boxplot of average beta values of all probes in TET2 WT and TET2 mutant AML patients (P=0.52). |
–
Furthermore, we have previously shown that genetic deficiency for Tet1 and Tet2 in mouse embryonic fibroblasts leads to specific hypermethylation of DNA methylation canyons [47]. Canyons are conserved regions of at least 3.5 kb that are largely depleted of DNA methylation and frequently harbor genes of developmental regulators. They are considerably larger than CpG islands and also contain CpG island shore and shelf regions [48]. Assuming that 2-HG inhibits TET enzymes, we expected canyons to be specifically targeted by hypermethylation in mIDH expressing AML patients. We used a high-coverage whole genome bisulfite sequencing dataset from an IDH WT AML patient [49] to identify 1711 canyons and 26,117 associated Infinium 450K probes. Average methylation ratios of these canyon-associated probes were only minimally (delta beta 0.014), but significantly increased in patients with mIDH (Fig. 4A). The small gain in methylation was evenly distributed over the length of the canyon and not enriched at canyon borders (Fig. 4B), which is distinct from Tet1/2-deficient cells [47]. These findings again support the notion that mIDH-associated genomic hypermethylation is mediated by factors other than TET inhibition.
–
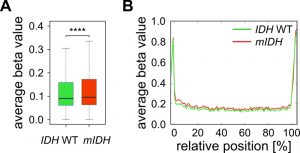 | FIGURE 4: TET-dependent DNA methylation canyons are not specifically affected by mIDH-associated hypermethylation in AML. (A) Average methylation ratios of canyon-associated probes in mIDH and IDH WT patients. The difference between the two groups was highly significant (**** P<0.0001). (B) Superposition of all size-normalized canyons depicting average methylation levels of these features in the two patient groups. |
–
D-2-hydroxyglutarate does not induce genomic hypermethylation in cancer cells
D-2-HG produced by mIDH enzymes has been shown to inhibit the hydroxylation activity of α-ketoglutarate dependent dioxygenases, including the TET enzymes [3][4]. However, these studies relied on 5hmC immunostainings upon TET overexpression or on in vitro enzymatic assays achieving only partial inhibition of TET activity, indicating that 2-HG is only a weak inhibitor of TET enzymes. Furthermore, the ability of D-2-HG to inhibit DNA demethylation in human cells has not been established yet. To mimic the conditions observed in mIDH AML patients, we incubated HL-60 cells in medium supplemented with a high concentration (30 mM) of synthetic D-2-HG. These cells had a significantly reduced proliferation rate (Fig. S5) but remained viable. Measurement of intracellular D-2-HG demonstrated that HL-60 cells imported the metabolite, resulting in intracellular concentrations that strongly exceeded those obtained by overexpression of mIDH2 (Fig. 5A). We next analyzed genomic DNA from cells treated with 30 mM of D-2-HG for three weeks in two independent experiments using Infinium methylation arrays. Comparison of D-2-HG treated with untreated HL-60 cells identified no significantly DMPs (Fig. 5B). Furthermore, the average methylation level was unchanged in treated compared to untreated cells (Fig. 5C; P=0.17). Thus, our data strongly suggest that D-2-HG is not sufficient to induce DNA hypermethylation in HL-60 cells. In addition, these findings confirm the notion that mIDH-associated hypermethylation is not caused by ineffective TET-mediated DNA demethylation.
–
 | FIGURE 5: D-2-HG does not induce genomic hypermethylation in cultured cells. (A) Measurement of intracellular D-2-HG concentrations using an enzymatic conversion assay [50]. HL-60 cells were cultured in medium containing 5 or 30 mM synthetic D-2-HG and quantifications of intracellular concentrations were performed at the indicated time points. The right panel shows intracellular concentrations in HL-60 cells transduced with empty vector (EV) or mIDH2. Statistical significance was calculated using the two-sided Student's t-test (*** P<0.001 and ** P<0.01). (B) Comparison of methylomes from D-2-HG treated (2-HG; 30 mM for 21 days) and untreated (u.t.) HL-60 cells by scatterplot. No significantly changed probes (adj. P<0.05) were found analyzing two biological replicates of each condition by EPIC methylation array. (C) Boxplot of average beta values of all probes in untreated and D-2-HG treated (30 mM for 21 days) HL-60 cells (P=0.17). |
–
mIDH-associated methylation patterns resemble myeloid progenitor methylomes
Methylome analysis of sorted hematopoietic cell types from various differentiation stages demonstrated that myeloid differentiation is associated with a global loss of DNA methylation [37][39]. AML samples are usually unpurified bone marrow aspirates with a high blast count comprising all kinds of hematopoietic differentiation stages. Since previous reports have shown that mIDH blocks differentiation [9][15][51][52][53], we reasoned that mIDH-associated genomic hypermethylation might simply reflect physiological methylation patterns of less differentiated myeloid progenitor cell types. We therefore analyzed the distribution of French–American–British (FAB) classes, which are routinely used to morphologically distinguish the predominant cell type and maturity of AML. This showed a clear enrichment of the relatively undifferentiated M1 subclass within the mIDH patients compared to the IDH WT cohort (Fig. 6A). Undifferentiated acute myeloblastic leukemia (M0) was also more frequent in the mIDH group than in the IDH WT group, while the remaining classes were relatively rare in mIDH patients or not present at all (Fig. 6A). Moreover, PCA showed that patients with a higher grade of maturation (M2 – M5) clustered together with the IDH WT patients (Fig. 6B), indicating that AML cells which manage to mature in the presence of IDH mutations adopt methylation profiles similar to IDH WT AML methylomes.
–
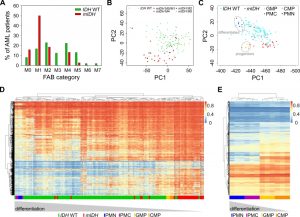 | FIGURE 6: mIDH-associated hypermethylation reflects enriched hypermethylated hematopoietic progenitor populations rather than a pathological hypermethylator phenotype. (A) Analysis of the distribution of the French–American–British (FAB) categories assigned in the AML dataset from TCGA within the IDH WT and mIDH patient groups. (B) PCA as in Fig. 1A. Different FAB categories in the mIDH patient group are colored as indicated. Variances captured by PC1 and PC2 were 17.3% and 8.3%, respectively. (C) PCA with 450K profiles from sorted human cells of four hematopoietic differentiation stages [38]. TCGA AML patient profiles of mIDH/IDH WT patients were projected on top of the clusters generated by analysis of the four cell types. CMP: common myeloid progenitor, GMP: granulocyte macrophage progenitor, PMC: promyelocyte, PMN: polymorphonuclear/terminally differentiated bone marrow neutrophil. (D) Heatmap of the 10,000 most differentially methylated probes between the CMP/GMP and PMC/PMN populations in the AML cohort of mIDH/IDH WT patients. Each row shows one probe and each column one patient. A hierarchical cluster dendrogram showing similarities between samples is depicted on the top. The color scale indicates beta values. (E) Heatmap and hierarchical cluster dendrogram of human hematopoietic cell types using the 10,000 most differentially methylated probes between mIDH and IDH WT AML patients. Each row shows one probe and each column one sample. The color scale indicates beta values. |
–
We also examined methylation patterns of four purified cell types of the human hematopoietic lineage (CMP, GMP, PMC and PMN), which were analyzed previously by 450K methylation arrays [38] and could therefore be directly integrated into our analysis. PCA segregated the less differentiated CD34 positive (CMP and GMP) from the more differentiated CD34 negative (PMC and PMN) samples. When AML methylation profiles were projected on top of these reference methylation profiles, mIDH samples clustered together with the undifferentiated cells, whereas IDH WT samples appeared closer to the differentiated cell types (Fig. 6C). Finally, we also subjected the 10,000 most differentially methylated probes between the progenitor cells (GMPs and CMPs) and the differentiated cells (PMCs and PMNs) to hierarchical clustering. This separated the two differentiation stages from each other and placed most mIDH AML patients in the same branch as the less differentiated myeloid progenitor cells (Fig. 6D). When we used the 10,000 most differentially methylated probes between mIDH and IDH WT patients to cluster the sorted hematopoietic cell types, CD34 positive progenitors were again separated from CD34 negative cells. Also the GMP and CMP samples were placed in different branches consistent with their differentiation state (Fig. 6E). Together, these findings identify physiological methylation states of arrested myeloid progenitor cells as a major source of mIDH-associated hypermethylation in AML.
–
DISCUSSION
DNA hypermethylation is a key feature of many cancer epigenomes but its origin and functional contribution to tumorigenesis remain controversial. Here, we have analyzed a large AML cohort from TCGA and found genomic hypermethylation associated with IDH1/2 mutations. This is consistent with previous studies and findings in other tumor entities such as lower-grade glioma or cholangiocarcinoma [15][21][54]. However, AML-specific hypermethylation patterns could not be identified in cells with mIDH2 overexpression or D-2-HG treatment. In conjunction with the paucity of large-scale DNA methylation changes in TET2 mutant AML patients, this critically challenges the current paradigm of mIDH-dependent DNA hypermethylation by D-2-HG-mediated TET inhibition.
–
The claim that mutant IDH induces hypermethylation through its action on TET enzymes [15][21], would suggest similar phenotypes upon expression of mIDH and loss of TET activity. However, in support of our study there are clinical differences in TET2 and IDH mutant diseases and distinct hematopoietic phenotypes in Tet2-deficient and mIdh expressing mice. Tet2-deficient mice show a more pronounced expansion of the stem cell pool than mIdh1 expressing animals and additionally display augmented repopulation activity and skewed differentiation [7][10][55][56][57]. Moreover, the effect of TET2 mutations on DNA methylation in human myeloid malignancies remains unclear with some studies identifying hypermethylation [15][45][58][59] and others hypomethylation [44][46]. Also, global 5mC levels were not affected in human erythropoietic cell types upon TET2/TET3 knockdown [60]. This is in agreement with our findings and suggests that TET deficiency and IDH mutations have distinct epigenetic effects in myeloid malignancies.
–
While the reported paracrine activity of 2-HG [33] should allow inhibition of TET activity without mIDH, DNA hypermethylation was not detectable in our cellular model. This indicates that hypermethylation may be induced by pathways that are independent of the catalytic activity of mIDH. It should be noted that AML cells with mIDH may be exposed to high 2-HG concentrations over a much longer period than several weeks and that they may also be influenced by 2-HG-mediated changes of the stromal niche [61]. However, if TET-mediated DNA demethylation was inhibited over several cell divisions, our in vitro model should display DNA hypermethylation. Further clarification may require longer 2-HG exposure or appropriate mouse models. Indeed, when 2-HG was administered to different leukemia mouse models over 4 weeks, leukemia-promoting effects were observed, but the metabolite was not sufficient to induce leukemia or change DNA methylation patterns to mimick IDH1 mutants [33]. Also, several reports have shown that the inhibition of D-2-HG production by mIDH inhibitors did not revert global DNA hypermethylation in glioma and AML cells [22][23][26]. These observations emphasize that depletion of 2-HG is not sufficient to restore normal methylomes.
–
An mIDH-dependent differentiation block has repeatedly been reported in different in vitro and in vivo systems [9][10][15][51][52][53]. Accordingly, we found an enrichment of FAB categories representing early differentiation stages in mIDH AML patients. Furthermore, our comparison with sorted myeloid cell types indicated that mIDH-associated methylation patterns strongly resemble myeloid progenitor cell types. We therefore propose that mIDH expression keeps AML cells in a less differentiated state that is reflected in their methylation landscape. IDH mutations are early events in the transformation of blood cells as suggested by analyses of premalignant cells and hematopoietic disorders [1][62][63][64]. mIDH may thus inhibit differentiation of the mutated progenitor and all clonally derived cells, whereas cells with a wildtype IDH allele may retain the capacity to differentiate further. Together with the finding that the TET inhibitor L-2-HG did not recapitulate the differentiation block observed with D-2-HG in cell-based experiments [9], our results suggest a TET-independent mechanism leading to differentiation arrest [3][51][65].
–
CpG island hypermethylation or more specifically CIMP has been described in many cancers, however it cannot always be correlated with IDH or other mutations [66][67][68][69]. Furthermore, methylation patterns of B-cell chronic lymphocytic leukemia were described to be highly related to normal methylomes of various differentiation stages [70][71][72]. In addition, CIMP was recently described as a cancer-independent feature of proliferating myeloid cells [73], which further challenged the cancer-specificity of this phenotype. Our findings suggest that mIDH-associated hypermethylation in AML reflects the relatively undifferentiated state of the cancer cells, rather than impaired DNA demethylation. Our study thereby furthers the understanding of mIDH-associated alterations in DNA methylation and emphasizes the need for a detailed characterization of the 2-HG-dependent and independent functions of mutant IDH enzymes.
MATERIALS AND METHODS
Patient data
Raw .IDAT Illumina Infinium 450K methylation array files, clinical patient parameters including FAB categories and mutational status were downloaded from the TCGA database or GDC Legacy Archive [20].
–
Cell culture, generation of stable mIDH2 expressing HL-60 cells and proliferation analysis
HL-60 cells were cultured in RPMI supplemented with 10% (v/v) fetal bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin. For generation of stable cell lines IDH2 R140Q was cloned into pLVX-IRES-ZsGreen1. HEK293T cells were grown to 90% confluency in 6-well plates and transfected with 2.5 µg of DNA mix consisting of pLVX-IRES-ZsGreen1 (empty vector)/pLVX-IRES-ZsGreen1-mIDH2, psPAX2 and pMD2.G in a ratio of 5:4:1 using lipofectamine 2000 according to the manufacturer´s instructions. After two days viral particles were harvested, filtered (0.45 µm) and 200,000 HL-60 cells/well were resuspended in virus-containing cell culture supernatant for infection. After a few days the top 30% of fluorescent cells were FACS sorted by ZsGreen1 expression to pools of at least 1000 cells. The sorting was repeated several times. For proliferation analysis, at each passage 500,000 cells were seeded in triplicates and after 3 – 4 days cell numbers/well were determined and cells were passaged. The population doubling level was computed from averages of triplicates as described before [74] and the cumulative population doubling level at each passage was plotted against time.
–
Cellular 2-HG treatment and quantification
D-2-HG (Sigma) was freshly dissolved in RPMI, the solution was rotated for 30 min at room temperature and sterile-filtrated (0.2 µm). 600,000 cells/well were seeded into a 24-well plate in 1 ml of D-2-HG supplemented medium. Medium was exchanged and cells were reduced to 600,000/well twice per week. Remaining cells were quickly washed in PBS for two times, pelleted and stored at -20°C. Intra- and extracellular D-2-HG quantifications were performed as described previously [50].
–
DNA and RNA isolation
Genomic DNA was isolated by lysing freshly prepared cell pellets in pre-lysis buffer (10 mM Tris-HCl (pH 8), 5 mM EDTA, 100 mM NaCl, 1.1% (v/v) SDS, 0.1 mg/ml Proteinase K and 0.04 mg/ml RNAse A) at 37°C. The next day proteins were precipitated by addition of 5 M NaCl and the DNA was isolated by isopropanol precipitation. Total RNA was isolated using TRIzol (Invitrogen).
–
cDNA synthesis and gene expression analysis
cDNA was synthesized from 1 µg RNA (QuantiTect Reverse Transcription Kit; Qiagen) following the manufacturer’s instructions. Quantitative RT-PCRs were performed in triplicate using Mesa green qPCR mastermix PLUS (Eurogentec) and the Lightcycler 480 Real-Time PCR system (Roche). Expression values were calculated from raw Ct values using the ΔΔCt method and ACTB as a reference gene.
–
Dotblots and Infinium EPIC methylation assay
Genomic DNA was isolated and 4 µg were used for dotblots as described before [47]. For methylation profiling 1 µg of DNA was used. The Infinium MethylationEPIC BeadChip assay was performed by the microarray unit of the DKFZ Genomics and Proteomics Core Facility. Briefly, per experiment and condition two biological replicates were used. Matched untreated or empty vector transduced cells served as controls. 500 ng of high quality genomic DNA were bisulfite converted using the EZ-96 DNA Methylation Kit (Zymo Research) according to the manufacturer’s instructions, whole genome amplified, enzymatically fragmented and denatured following the recommendations of the Infinium HD Assay Methylation Protocol Guide (Illumina), and hybridized to the BeadChip.
–
Infinium analysis pipeline
The raw intensity data files were normalized, quality-filtered and statistically analyzed as described before [75]. In brief, the minfi package [41] was used to load IDAT files into R. Data were normalized with the SWAN method [76] without prior background correction. Cross-reactive and SNP containing probes [77], probes on the sex chromosomes and probes with a low detection P-value (>0.01) were omitted. In order to allow comparisons between 450K and EPIC datasets only probes present on both chips were included. PCA graphs were generated by the plotPCA function provided by the R affycoretools package [78] with all probes used to compute principal components. Heatmaps were created by the heatmap.2 function of the R gplots package [79]. For clustering the euclidean distance function was used to compute the distance matrix and the complete linkage method to obtain hierarchical clustering. The RColorBrewer package was used to create the assigned colors [80].
–
Mapping of canyons and canyon analysis using 450K data
Canyons were computed as described before [47] using a whole genome bisulfite sequencing dataset from a female AML patient with wildtype IDH genes available from the Blueprint Epigenome Consortium (http://www.blueprint-epigenome.eu; [49]). This led to a set of 1,711 canyons covering a total of 26,117 Infinium 450K probes. For further analysis the canyons were size-normalized and the average methylation profiles of IDH WT and mIDH samples were computed.
–
Integrated Principal Component Analysis of FANTOM and TCGA data
A PCA of the 450K methylation data of 12 differentiation related blood cell datasets generated as a part of the FANTOM5 project [38] was computed using the R package FactoMineR (http://factominer.free.fr). TCGA AML patient methylation profiles of mIDH/IDH WT patients were projected onto this PCA using the new basis vectors computed by the PCA.
–
Heatmaps and cluster dendrograms
Heatmaps and cluster dendrograms were computed using the R package pheatmap.
–
Statistical methods
Differentially methylated probes between wildtype and mutant patients or HL-60 cells were identified by a similar approach as implemented in minfi [41]. Briefly, differentially methylated CpGs were detected by a linear model fit of methylation values, which enabled an empirical Bayes moderation of standard errors from estimated methylation differences. Subsequently, P-values were adjusted for false discovery rate using the Benjamini-Hochberg procedure. Probes with an adjusted P-value (adj. P) <0.05 were considered as differentially methylated. Significance of alterations in global methylation was inferred by a two-sided, unpaired, Welch-corrected t-test. Gene expression and D-2-HG differences were tested using an unpaired, two-sided Student’s t-test or Welch Two Sample t-test if variances were unequal. A hypergeometric test was used to calculate the significance level of overlapping probes.
–
Data access
Infinium raw data (.IDAT files) of HL-60 cells have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-6059.
References
- L. Dang, K. Yen, and E. Attar, "IDH mutations in cancer and progress toward development of targeted therapeutics", Annals of Oncology, vol. 27, pp. 599-608, 2016. http://dx.doi.org/10.1093/annonc/mdw013
- L. Dang, D.W. White, S. Gross, B.D. Bennett, M.A. Bittinger, E.M. Driggers, V.R. Fantin, H.G. Jang, S. Jin, M.C. Keenan, K.M. Marks, R.M. Prins, P.S. Ward, K.E. Yen, L.M. Liau, J.D. Rabinowitz, L.C. Cantley, C.B. Thompson, M.G. Vander Heiden, and S.M. Su, "Cancer-associated IDH1 mutations produce 2-hydroxyglutarate", Nature, vol. 462, pp. 739-744, 2009. http://dx.doi.org/10.1038/nature08617
- W. Xu, H. Yang, Y. Liu, Y. Yang, P. Wang, S. Kim, S. Ito, C. Yang, P. Wang, M. Xiao, L. Liu, W. Jiang, J. Liu, J. Zhang, B. Wang, S. Frye, Y. Zhang, Y. Xu, Q. Lei, K. Guan, S. Zhao, and Y. Xiong, "Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases", Cancer Cell, vol. 19, pp. 17-30, 2011. http://dx.doi.org/10.1016/j.ccr.2010.12.014
- P. Koivunen, S. Lee, C.G. Duncan, G. Lopez, G. Lu, S. Ramkissoon, J.A. Losman, P. Joensuu, U. Bergmann, S. Gross, J. Travins, S. Weiss, R. Looper, K.L. Ligon, R.G.W. Verhaak, H. Yan, and W.G. Kaelin Jr, "Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation", Nature, vol. 483, pp. 484-488, 2012. http://dx.doi.org/10.1038/nature10898
- S. Zhao, Y. Lin, W. Xu, W. Jiang, Z. Zha, P. Wang, W. Yu, Z. Li, L. Gong, Y. Peng, J. Ding, Q. Lei, K. Guan, and Y. Xiong, "Glioma-Derived Mutations in IDH1 Dominantly Inhibit IDH1 Catalytic Activity and Induce HIF-1α", Science, vol. 324, pp. 261-265, 2009. http://dx.doi.org/10.1126/science.1170944
- S.M. Chan, D. Thomas, M.R. Corces-Zimmerman, S. Xavy, S. Rastogi, W. Hong, F. Zhao, B.C. Medeiros, D.A. Tyvoll, and R. Majeti, "Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia", Nature Medicine, vol. 21, pp. 178-184, 2015. http://dx.doi.org/10.1038/nm.3788
- S. Inoue, W.Y. Li, A. Tseng, I. Beerman, A.J. Elia, S.C. Bendall, F. Lemonnier, K.J. Kron, D.W. Cescon, Z. Hao, E.F. Lind, N. Takayama, A.C. Planello, S.Y. Shen, A.H. Shih, D.M. Larsen, Q. Li, B.E. Snow, A. Wakeham, J. Haight, C. Gorrini, C. Bassi, K.L. Thu, K. Murakami, A.R. Elford, T. Ueda, K. Straley, K.E. Yen, G. Melino, L. Cimmino, I. Aifantis, R.L. Levine, D.D. De Carvalho, M. Lupien, D.J. Rossi, G.P. Nolan, R.A. Cairns, and T.W. Mak, "Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2", Cancer Cell, vol. 30, pp. 337-348, 2016. http://dx.doi.org/10.1016/j.ccell.2016.05.018
- F. Li, X. He, D. Ye, Y. Lin, H. Yu, C. Yao, L. Huang, J. Zhang, F. Wang, S. Xu, X. Wu, L. Liu, C. Yang, J. Shi, X. He, J. Liu, Y. Qu, F. Guo, J. Zhao, W. Xu, and S. Zhao, "NADP+-IDH Mutations Promote Hypersuccinylation that Impairs Mitochondria Respiration and Induces Apoptosis Resistance", Molecular Cell, vol. 60, pp. 661-675, 2015. http://dx.doi.org/10.1016/j.molcel.2015.10.017
- J. Losman, R.E. Looper, P. Koivunen, S. Lee, R.K. Schneider, C. McMahon, G.S. Cowley, D.E. Root, B.L. Ebert, and W.G. Kaelin, "( R )-2-Hydroxyglutarate Is Sufficient to Promote Leukemogenesis and Its Effects Are Reversible", Science, vol. 339, pp. 1621-1625, 2013. http://dx.doi.org/10.1126/science.1231677
- M. Sasaki, C.B. Knobbe, J.C. Munger, E.F. Lind, D. Brenner, A. Brüstle, I.S. Harris, R. Holmes, A. Wakeham, J. Haight, A. You-Ten, W.Y. Li, S. Schalm, S.M. Su, C. Virtanen, G. Reifenberger, P.S. Ohashi, D.L. Barber, M.E. Figueroa, A. Melnick, J. Zúñiga-Pflücker, and T.W. Mak, "IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics", Nature, vol. 488, pp. 656-659, 2012. http://dx.doi.org/10.1038/nature11323
- M. Sasaki, C.B. Knobbe, M. Itsumi, A.J. Elia, I.S. Harris, I.I.C. Chio, R.A. Cairns, S. McCracken, A. Wakeham, J. Haight, A.Y. Ten, B. Snow, T. Ueda, S. Inoue, K. Yamamoto, M. Ko, A. Rao, K.E. Yen, S.M. Su, and T.W. Mak, "D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function", Genes & Development, vol. 26, pp. 2038-2049, 2012. http://dx.doi.org/10.1101/gad.198200.112
- P.A. Jones, and S.B. Baylin, "The Epigenomics of Cancer", Cell, vol. 128, pp. 683-692, 2007. http://dx.doi.org/10.1016/j.cell.2007.01.029
- A. Akalin, F.E. Garrett-Bakelman, M. Kormaksson, J. Busuttil, L. Zhang, I. Khrebtukova, T.A. Milne, Y. Huang, D. Biswas, J.L. Hess, C.D. Allis, R.G. Roeder, P.J.M. Valk, B. Löwenberg, R. Delwel, H.F. Fernandez, E. Paietta, M.S. Tallman, G.P. Schroth, C.E. Mason, A. Melnick, and M.E. Figueroa, "Base-Pair Resolution DNA Methylation Sequencing Reveals Profoundly Divergent Epigenetic Landscapes in Acute Myeloid Leukemia", PLoS Genetics, vol. 8, pp. e1002781, 2012. http://dx.doi.org/10.1371/journal.pgen.1002781
- C.G. Duncan, B.G. Barwick, G. Jin, C. Rago, P. Kapoor-Vazirani, D.R. Powell, J. Chi, D.D. Bigner, P.M. Vertino, and H. Yan, "A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation", Genome Research, vol. 22, pp. 2339-2355, 2012. http://dx.doi.org/10.1101/gr.132738.111
- M.E. Figueroa, O. Abdel-Wahab, C. Lu, P.S. Ward, J. Patel, A. Shih, Y. Li, N. Bhagwat, A. Vasanthakumar, H.F. Fernandez, M.S. Tallman, Z. Sun, K. Wolniak, J.K. Peeters, W. Liu, S.E. Choe, V.R. Fantin, E. Paietta, B. Löwenberg, J.D. Licht, L.A. Godley, R. Delwel, P.J. Valk, C.B. Thompson, R.L. Levine, and A. Melnick, "Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation", Cancer Cell, vol. 18, pp. 553-567, 2010. http://dx.doi.org/10.1016/j.ccr.2010.11.015
- W.A. Flavahan, Y. Drier, B.B. Liau, S.M. Gillespie, A.S. Venteicher, A.O. Stemmer-Rachamimov, M.L. Suvà, and B.E. Bernstein, "Insulator dysfunction and oncogene activation in IDH mutant gliomas", Nature, vol. 529, pp. 110-114, 2015. http://dx.doi.org/10.1038/nature16490
- A. Kernytsky, F. Wang, E. Hansen, S. Schalm, K. Straley, C. Gliser, H. Yang, J. Travins, S. Murray, M. Dorsch, S. Agresta, D.P. Schenkein, S.A. Biller, S.M. Su, W. Liu, and K.E. Yen, "IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition", Blood, vol. 125, pp. 296-303, 2015. http://dx.doi.org/10.1182/blood-2013-10-533604
- D. Sturm, H. Witt, V. Hovestadt, D. Khuong-Quang, D. Jones, C. Konermann, E. Pfaff, M. Tönjes, M. Sill, S. Bender, M. Kool, M. Zapatka, N. Becker, M. Zucknick, T. Hielscher, X. Liu, A. Fontebasso, M. Ryzhova, S. Albrecht, K. Jacob, M. Wolter, M. Ebinger, M. Schuhmann, T. van Meter, M. Frühwald, H. Hauch, A. Pekrun, B. Radlwimmer, T. Niehues, G. von Komorowski, M. Dürken, A. Kulozik, J. Madden, A. Donson, N. Foreman, R. Drissi, M. Fouladi, W. Scheurlen, A. von Deimling, C. Monoranu, W. Roggendorf, C. Herold-Mende, A. Unterberg, C. Kramm, J. Felsberg, C. Hartmann, B. Wiestler, W. Wick, T. Milde, O. Witt, A. Lindroth, J. Schwartzentruber, D. Faury, A. Fleming, M. Zakrzewska, P. Liberski, K. Zakrzewski, P. Hauser, M. Garami, A. Klekner, L. Bognar, S. Morrissy, F. Cavalli, M. Taylor, P. van Sluis, J. Koster, R. Versteeg, R. Volckmann, T. Mikkelsen, K. Aldape, G. Reifenberger, V. Collins, J. Majewski, A. Korshunov, P. Lichter, C. Plass, N. Jabado, and S. Pfister, "Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma", Cancer Cell, vol. 22, pp. 425-437, 2012. http://dx.doi.org/10.1016/j.ccr.2012.08.024
- A. Chaturvedi, M.M. Araujo Cruz, N. Jyotsana, A. Sharma, H. Yun, K. Görlich, M. Wichmann, A. Schwarzer, M. Preller, F. Thol, J. Meyer, R. Haemmerle, E.A. Struys, E.E. Jansen, U. Modlich, Z. Li, L.M. Sly, R. Geffers, R. Lindner, D.J. Manstein, U. Lehmann, J. Krauter, A. Ganser, and M. Heuser, "Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML", Blood, vol. 122, pp. 2877-2887, 2013. http://dx.doi.org/10.1182/blood-2013-03-491571
- . , "Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia", New England Journal of Medicine, vol. 368, pp. 2059-2074, 2013. http://dx.doi.org/10.1056/NEJMoa1301689
- S. Turcan, D. Rohle, A. Goenka, L.A. Walsh, F. Fang, E. Yilmaz, C. Campos, A.W.M. Fabius, C. Lu, P.S. Ward, C.B. Thompson, A. Kaufman, O. Guryanova, R. Levine, A. Heguy, A. Viale, L.G.T. Morris, J.T. Huse, I.K. Mellinghoff, and T.A. Chan, "IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype", Nature, vol. 483, pp. 479-483, 2012. http://dx.doi.org/10.1038/nature10866
- S. Pusch, S. Krausert, V. Fischer, J. Balss, M. Ott, D. Schrimpf, D. Capper, F. Sahm, J. Eisel, A. Beck, M. Jugold, V. Eichwald, S. Kaulfuss, O. Panknin, H. Rehwinkel, K. Zimmermann, R.C. Hillig, J. Guenther, L. Toschi, R. Neuhaus, A. Haegebart, H. Hess-Stumpp, M. Bauser, W. Wick, A. Unterberg, C. Herold-Mende, M. Platten, and A. von Deimling, "Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo", Acta Neuropathologica, vol. 133, pp. 629-644, 2017. http://dx.doi.org/10.1007/s00401-017-1677-y
- A. Chaturvedi, L. Herbst, S. Pusch, L. Klett, R. Goparaju, D. Stichel, S. Kaulfuss, O. Panknin, K. Zimmermann, L. Toschi, R. Neuhaus, A. Haegebarth, H. Rehwinkel, H. Hess-Stumpp, M. Bauser, T. Bochtler, E.A. Struys, A. Sharma, A. Bakkali, R. Geffers, M.M. Araujo-Cruz, F. Thol, R. Gabdoulline, A. Ganser, A.D. Ho, A. von Deimling, K. Rippe, M. Heuser, and A. Krämer, "Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo", Leukemia, vol. 31, pp. 2020-2028, 2017. http://dx.doi.org/10.1038/leu.2017.46
- C. DiNardo, S. de Botton, D. Pollyea, E. Stein, A. Fathi, G. Roboz, R. Collins, R. Swords, I. Flinn, J. Altmann, M. Tallmann, H. Kantarjian, A. Derti, M. Goldwasser, M. Prahl, B. Wu, K. Yen, S. Agresta, and R. Stone, " Molecular Profiling and Relationship with Clinical Response in Patients with IDH1 Mutation-Positive Hematologic Malignancies Receiving AG-120, a First-in-Class Potent Inhibitor of Mutant IDH1.", Blood 126(1306), 2015.
- H. Burris, I. Mellinghoff, E. Maher, P. Wen, M. Beeram, M. Touat, J. Faris, N. Azad, T. Cloughesy, L. Gore, J. Trent, D.V. Hoff, M. Goldwasser, B. Fan, and S. Agresta, "Abstract PL04-05: The first reported results of AG-120, a first-in-class, potent inhibitor of the IDH1 mutant protein, in a Phase I study of patients with advanced IDH1-mutant solid tumors, including gliomas", Molecular Cancer Therapeutics, vol. 14, pp. PL04-05-PL04-05, 2015. http://dx.doi.org/10.1158/1535-7163.targ-15-pl04-05
- D. Rohle, J. Popovici-Muller, N. Palaskas, S. Turcan, C. Grommes, C. Campos, J. Tsoi, O. Clark, B. Oldrini, E. Komisopoulou, K. Kunii, A. Pedraza, S. Schalm, L. Silverman, A. Miller, F. Wang, H. Yang, Y. Chen, A. Kernytsky, M.K. Rosenblum, W. Liu, S.A. Biller, S.M. Su, C.W. Brennan, T.A. Chan, T.G. Graeber, K.E. Yen, and I.K. Mellinghoff, "An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells", Science, vol. 340, pp. 626-630, 2013. http://dx.doi.org/10.1126/science.1236062
- F. Wang, J. Travins, B. DeLaBarre, V. Penard-Lacronique, S. Schalm, E. Hansen, K. Straley, A. Kernytsky, W. Liu, C. Gliser, H. Yang, S. Gross, E. Artin, V. Saada, E. Mylonas, C. Quivoron, J. Popovici-Muller, J.O. Saunders, F.G. Salituro, S. Yan, S. Murray, W. Wei, Y. Gao, L. Dang, M. Dorsch, S. Agresta, D.P. Schenkein, S.A. Biller, S.M. Su, S. de Botton, and K.E. Yen, "Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation", Science, vol. 340, pp. 622-626, 2013. http://dx.doi.org/10.1126/science.1234769
- J. Popovici-Muller, J.O. Saunders, F.G. Salituro, J.M. Travins, S. Yan, F. Zhao, S. Gross, L. Dang, K.E. Yen, H. Yang, K.S. Straley, S. Jin, K. Kunii, V.R. Fantin, S. Zhang, Q. Pan, D. Shi, S.A. Biller, and S.M. Su, "Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo", ACS Medicinal Chemistry Letters, vol. 3, pp. 850-855, 2012. http://dx.doi.org/10.1021/ml300225h
- U.C. Okoye-Okafor, B. Bartholdy, J. Cartier, E.N. Gao, B. Pietrak, A.R. Rendina, C. Rominger, C. Quinn, A. Smallwood, K.J. Wiggall, A.J. Reif, S.J. Schmidt, H. Qi, H. Zhao, G. Joberty, M. Faelth-Savitski, M. Bantscheff, G. Drewes, C. Duraiswami, P. Brady, A. Groy, S. Narayanagari, I. Antony-Debre, K. Mitchell, H.R. Wang, Y. Kao, M. Christopeit, L. Carvajal, L. Barreyro, E. Paietta, H. Makishima, B. Will, N. Concha, N.D. Adams, B. Schwartz, M.T. McCabe, J. Maciejewski, A. Verma, and U. Steidl, "New IDH1 mutant inhibitors for treatment of acute myeloid leukemia", Nature Chemical Biology, vol. 11, pp. 878-886, 2015. http://dx.doi.org/10.1038/nchembio.1930
- G. Deng, J. Shen, M. Yin, J. McManus, M. Mathieu, P. Gee, T. He, C. Shi, O. Bedel, L.R. McLean, F. Le-Strat, Y. Zhang, J. Marquette, Q. Gao, B. Zhang, A. Rak, D. Hoffmann, E. Rooney, A. Vassort, W. Englaro, Y. Li, V. Patel, F. Adrian, S. Gross, D. Wiederschain, H. Cheng, and S. Licht, "Selective Inhibition of Mutant Isocitrate Dehydrogenase 1 (IDH1) via Disruption of a Metal Binding Network by an Allosteric Small Molecule", Journal of Biological Chemistry, vol. 290, pp. 762-774, 2015. http://dx.doi.org/10.1074/jbc.M114.608497
- K. Yen, J. Travins, F. Wang, M.D. David, E. Artin, K. Straley, A. Padyana, S. Gross, B. DeLaBarre, E. Tobin, Y. Chen, R. Nagaraja, S. Choe, L. Jin, Z. Konteatis, G. Cianchetta, J.O. Saunders, F.G. Salituro, C. Quivoron, P. Opolon, O. Bawa, V. Saada, A. Paci, S. Broutin, O.A. Bernard, S. de Botton, B.S. Marteyn, M. Pilichowska, Y. Xu, C. Fang, F. Jiang, W. Wei, S. Jin, L. Silverman, W. Liu, H. Yang, L. Dang, M. Dorsch, V. Penard-Lacronique, S.A. Biller, and S.M. Su, "AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations", Cancer Discovery, vol. 7, pp. 478-493, 2017. http://dx.doi.org/10.1158/2159-8290.CD-16-1034
- A.H. Shih, C. Meydan, K. Shank, F.E. Garrett-Bakelman, P.S. Ward, A.M. Intlekofer, A. Nazir, E.M. Stein, K. Knapp, J. Glass, J. Travins, K. Straley, C. Gliser, C.E. Mason, K. Yen, C.B. Thompson, A. Melnick, and R.L. Levine, "Combination Targeted Therapy to Disrupt Aberrant Oncogenic Signaling and Reverse Epigenetic Dysfunction in IDH2- and TET2-Mutant Acute Myeloid Leukemia", Cancer Discovery, vol. 7, pp. 494-505, 2017. http://dx.doi.org/10.1158/2159-8290.CD-16-1049
- A. Chaturvedi, M.M. Araujo Cruz, N. Jyotsana, A. Sharma, R. Goparaju, A. Schwarzer, K. Görlich, R. Schottmann, E.A. Struys, E.E. Jansen, C. Rohde, C. Müller-Tidow, R. Geffers, G. Göhring, A. Ganser, F. Thol, and M. Heuser, "Enantiomer-specific and paracrine leukemogenicity of mutant IDH metabolite 2-hydroxyglutarate", Leukemia, vol. 30, pp. 1708-1715, 2016. http://dx.doi.org/10.1038/leu.2016.71
- Z.D. Smith, and A. Meissner, "DNA methylation: roles in mammalian development", Nature Reviews Genetics, vol. 14, pp. 204-220, 2013. http://dx.doi.org/10.1038/nrg3354
- E.A. Houseman, S. Kim, K.T. Kelsey, and J.K. Wiencke, "DNA Methylation in Whole Blood: Uses and Challenges", Current Environmental Health Reports, vol. 2, pp. 145-154, 2015. http://dx.doi.org/10.1007/s40572-015-0050-3
- H. Ji, L.I.R. Ehrlich, J. Seita, P. Murakami, A. Doi, P. Lindau, H. Lee, M.J. Aryee, R.A. Irizarry, K. Kim, D.J. Rossi, M.A. Inlay, T. Serwold, H. Karsunky, L. Ho, G.Q. Daley, I.L. Weissman, and A.P. Feinberg, "Comprehensive methylome map of lineage commitment from haematopoietic progenitors", Nature, vol. 467, pp. 338-342, 2010. http://dx.doi.org/10.1038/nature09367
- M. Farlik, F. Halbritter, F. Müller, F.A. Choudry, P. Ebert, J. Klughammer, S. Farrow, A. Santoro, V. Ciaurro, A. Mathur, R. Uppal, H.G. Stunnenberg, W.H. Ouwehand, E. Laurenti, T. Lengauer, M. Frontini, and C. Bock, "DNA Methylation Dynamics of Human Hematopoietic Stem Cell Differentiation", Cell Stem Cell, vol. 19, pp. 808-822, 2016. http://dx.doi.org/10.1016/j.stem.2016.10.019
- M. Rönnerblad, R. Andersson, T. Olofsson, I. Douagi, M. Karimi, S. Lehmann, I. Hoof, M. de Hoon, M. Itoh, S. Nagao-Sato, H. Kawaji, T. Lassmann, P. Carninci, Y. Hayashizaki, A.R.R. Forrest, A. Sandelin, K. Ekwall, E. Arner, and A. Lennartsson, "Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation", Blood, vol. 123, pp. e79-e89, 2014. http://dx.doi.org/10.1182/blood-2013-02-482893
- C. Bock, I. Beerman, W. Lien, Z. Smith, H. Gu, P. Boyle, A. Gnirke, E. Fuchs, D. Rossi, and A. Meissner, "DNA Methylation Dynamics during In Vivo Differentiation of Blood and Skin Stem Cells", Molecular Cell, vol. 47, pp. 633-647, 2012. http://dx.doi.org/10.1016/j.molcel.2012.06.019
- E. Passegué, C.H.M. Jamieson, L.E. Ailles, and I.L. Weissman, "Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics?", Proceedings of the National Academy of Sciences, vol. 100, pp. 11842-11849, 2003. http://dx.doi.org/10.1073/pnas.2034201100
- M.J. Aryee, A.E. Jaffe, H. Corrada-Bravo, C. Ladd-Acosta, A.P. Feinberg, K.D. Hansen, and R.A. Irizarry, "Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays", Bioinformatics, vol. 30, pp. 1363-1369, 2014. http://dx.doi.org/10.1093/bioinformatics/btu049
- D. Gaidatzis, L. Burger, R. Murr, A. Lerch, S. Dessus-Babus, D. Schübeler, and M.B. Stadler, "DNA Sequence Explains Seemingly Disordered Methylation Levels in Partially Methylated Domains of Mammalian Genomes", PLoS Genetics, vol. 10, pp. e1004143, 2014. http://dx.doi.org/10.1371/journal.pgen.1004143
- L. Guelen, L. Pagie, E. Brasset, W. Meuleman, M.B. Faza, W. Talhout, B.H. Eussen, A. de Klein, L. Wessels, W. de Laat, and B. van Steensel, "Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions", Nature, vol. 453, pp. 948-951, 2008. http://dx.doi.org/10.1038/nature06947
- M. Ko, Y. Huang, A.M. Jankowska, U.J. Pape, M. Tahiliani, H.S. Bandukwala, J. An, E.D. Lamperti, K.P. Koh, R. Ganetzky, X.S. Liu, L. Aravind, S. Agarwal, J.P. Maciejewski, and A. Rao, "Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2", Nature, vol. 468, pp. 839-843, 2010. http://dx.doi.org/10.1038/nature09586
- J. Yamazaki, R. Taby, A. Vasanthakumar, T. Macrae, K.R. Ostler, L. Shen, H.M. Kantarjian, M.R. Estecio, J. Jelinek, L.A. Godley, and J.J. Issa, "Effects ofTET2mutations on DNA methylation in chronic myelomonocytic leukemia", Epigenetics, vol. 7, pp. 201-207, 2012. http://dx.doi.org/10.4161/epi.7.2.19015
- C. Pérez, N. Martínez-Calle, J.I. Martín-Subero, V. Segura, E. Delabesse, M. Fernandez-Mercado, L. Garate, S. Alvarez, J. Rifon, S. Varea, J. Boultwood, J.S. Wainscoat, J.C. Cigudosa, M.J. Calasanz, N.C.P. Cross, F. Prósper, and X. Agirre, "TET2 Mutations Are Associated with Specific 5-Methylcytosine and 5-Hydroxymethylcytosine Profiles in Patients with Chronic Myelomonocytic Leukemia", PLoS ONE, vol. 7, pp. e31605, 2012. http://dx.doi.org/10.1371/journal.pone.0031605
- L. Wiehle, G. Raddatz, T. Musch, M.M. Dawlaty, R. Jaenisch, F. Lyko, and A. Breiling, "Tet1 and Tet2 Protect DNA Methylation Canyons against Hypermethylation", Molecular and Cellular Biology, vol. 36, pp. 452-461, 2016. http://dx.doi.org/10.1128/MCB.00587-15
- M. Jeong, D. Sun, M. Luo, Y. Huang, G.A. Challen, B. Rodriguez, X. Zhang, L. Chavez, H. Wang, R. Hannah, S. Kim, L. Yang, M. Ko, R. Chen, B. Göttgens, J. Lee, P. Gunaratne, L.A. Godley, G.J. Darlington, A. Rao, W. Li, and M.A. Goodell, "Large conserved domains of low DNA methylation maintained by Dnmt3a", Nature Genetics, vol. 46, pp. 17-23, 2013. http://dx.doi.org/10.1038/ng.2836
- R. Schuyler, A. Merkel, E. Raineri, L. Altucci, E. Vellenga, J. Martens, F. Pourfarzad, T. Kuijpers, F. Burden, S. Farrow, K. Downes, W. Ouwehand, L. Clarke, A. Datta, E. Lowy, P. Flicek, M. Frontini, H. Stunnenberg, J. Martín-Subero, I. Gut, and S. Heath, "Distinct Trends of DNA Methylation Patterning in the Innate and Adaptive Immune Systems", Cell Reports, vol. 17, pp. 2101-2111, 2016. http://dx.doi.org/10.1016/j.celrep.2016.10.054
- J. Balss, S. Pusch, A. Beck, C. Herold-Mende, A. Krämer, C. Thiede, W. Buckel, C. Langhans, J.G. Okun, and A. von Deimling, "Enzymatic assay for quantitative analysis of (d)-2-hydroxyglutarate", Acta Neuropathologica, vol. 124, pp. 883-891, 2012. http://dx.doi.org/10.1007/s00401-012-1060-y
- C. Lu, P.S. Ward, G.S. Kapoor, D. Rohle, S. Turcan, O. Abdel-Wahab, C.R. Edwards, R. Khanin, M.E. Figueroa, A. Melnick, K.E. Wellen, D.M. O’Rourke, S.L. Berger, T.A. Chan, R.L. Levine, I.K. Mellinghoff, and C.B. Thompson, "IDH mutation impairs histone demethylation and results in a block to cell differentiation", Nature, vol. 483, pp. 474-478, 2012. http://dx.doi.org/10.1038/nature10860
- Y. Jin, H. Elalaf, M. Watanabe, S. Tamaki, S. Hineno, K. Matsunaga, K. Woltjen, Y. Kobayashi, S. Nagata, M. Ikeya, T. Kato, T. Okamoto, S. Matsuda, and J. Toguchida, "Mutant IDH1 Dysregulates the Differentiation of Mesenchymal Stem Cells in Association with Gene-Specific Histone Modifications to Cartilage- and Bone-Related Genes", PLOS ONE, vol. 10, pp. e0131998, 2015. http://dx.doi.org/10.1371/journal.pone.0131998
- S.K. Saha, C.A. Parachoniak, K.S. Ghanta, J. Fitamant, K.N. Ross, M.S. Najem, S. Gurumurthy, E.A. Akbay, D. Sia, H. Cornella, O. Miltiadous, C. Walesky, V. Deshpande, A.X. Zhu, A.F. Hezel, K.E. Yen, K.S. Straley, J. Travins, J. Popovici-Muller, C. Gliser, C.R. Ferrone, U. Apte, J.M. Llovet, K. Wong, S. Ramaswamy, and N. Bardeesy, "Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer", Nature, vol. 513, pp. 110-114, 2014. http://dx.doi.org/10.1038/nature13441
- P. Wang, Q. Dong, C. Zhang, P. Kuan, Y. Liu, W.R. Jeck, J.B. Andersen, W. Jiang, G.L. Savich, T. Tan, J.T. Auman, J.M. Hoskins, A.D. Misher, C.D. Moser, S.M. Yourstone, J.W. Kim, K. Cibulskis, G. Getz, H.V. Hunt, S.S. Thorgeirsson, L.R. Roberts, D. Ye, K. Guan, Y. Xiong, L. Qin, and D.Y. Chiang, "Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas", Oncogene, vol. 32, pp. 3091-3100, 2012. http://dx.doi.org/10.1038/onc.2012.315
- Z. Li, X. Cai, C. Cai, J. Wang, W. Zhang, B.E. Petersen, F. Yang, and M. Xu, "Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies", Blood, vol. 118, pp. 4509-4518, 2011. http://dx.doi.org/10.1182/blood-2010-12-325241
- J. Losman, and W.G. Kaelin, "What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer", Genes & Development, vol. 27, pp. 836-852, 2013. http://dx.doi.org/10.1101/gad.217406.113
- K. Moran-Crusio, L. Reavie, A. Shih, O. Abdel-Wahab, D. Ndiaye-Lobry, C. Lobry, M. Figueroa, A. Vasanthakumar, J. Patel, X. Zhao, F. Perna, S. Pandey, J. Madzo, C. Song, Q. Dai, C. He, S. Ibrahim, M. Beran, J. Zavadil, S. Nimer, A. Melnick, L. Godley, I. Aifantis, and R. Levine, "Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation", Cancer Cell, vol. 20, pp. 11-24, 2011. http://dx.doi.org/10.1016/j.ccr.2011.06.001
- K.D. Rasmussen, G. Jia, J.V. Johansen, M.T. Pedersen, N. Rapin, F.O. Bagger, B.T. Porse, O.A. Bernard, J. Christensen, and K. Helin, "Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis", Genes & Development, vol. 29, pp. 910-922, 2015. http://dx.doi.org/10.1101/gad.260174.115
- J. Yamazaki, J. Jelinek, Y. Lu, M. Cesaroni, J. Madzo, F. Neumann, R. He, R. Taby, A. Vasanthakumar, T. Macrae, K.R. Ostler, H.M. Kantarjian, S. Liang, M.R. Estecio, L.A. Godley, and J.J. Issa, "TET2 Mutations Affect Non-CpG Island DNA Methylation at Enhancers and Transcription Factor–Binding Sites in Chronic Myelomonocytic Leukemia", Cancer Research, vol. 75, pp. 2833-2843, 2015. http://dx.doi.org/10.1158/0008-5472.CAN-14-0739
- H. Yan, Y. Wang, X. Qu, J. Li, J. Hale, Y. Huang, C. An, J. Papoin, X. Guo, L. Chen, Q. Kang, W. Li, V.P. Schulz, P.G. Gallagher, C.D. Hillyer, N. Mohandas, and X. An, "Distinct roles for TET family proteins in regulating human erythropoiesis", Blood, vol. 129, pp. 2002-2012, 2017. http://dx.doi.org/10.1182/blood-2016-08-736587
- J. Chen, Y. Lai, H. Tsai, C. Kuo, B.L. Yen, S. Yeh, H.S. Sun, and W. Hung, "The oncometabolite R-2-hydroxyglutarate activates NF-κB-dependent tumor-promoting stromal niche for acute myeloid leukemia cells", Scientific Reports, vol. 6, 2016. http://dx.doi.org/10.1038/srep32428
- L.I. Shlush, S. Zandi, A. Mitchell, W.C. Chen, J.M. Brandwein, V. Gupta, J.A. Kennedy, A.D. Schimmer, A.C. Schuh, K.W. Yee, J.L. McLeod, M. Doedens, J.J.F. Medeiros, R. Marke, H.J. Kim, K. Lee, J.D. McPherson, T.J. Hudson, T.H. Pan-Leukemia Gene Panel Consortium, A.M.K. Brown, F. Yousif, Q.M. Trinh, L.D. Stein, M.D. Minden, J.C.Y. Wang, and J.E. Dick, "Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia", Nature, vol. 506, pp. 328-333, 2014. http://dx.doi.org/10.1038/nature13038
- M.R. Corces-Zimmerman, W. Hong, I.L. Weissman, B.C. Medeiros, and R. Majeti, "Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission", Proceedings of the National Academy of Sciences, vol. 111, pp. 2548-2553, 2014. http://dx.doi.org/10.1073/pnas.1324297111
- J. Welch, T. Ley, D. Link, C. Miller, D. Larson, D. Koboldt, L. Wartman, T. Lamprecht, F. Liu, J. Xia, C. Kandoth, R. Fulton, M. McLellan, D. Dooling, J. Wallis, K. Chen, C. Harris, H. Schmidt, J. Kalicki-Veizer, C. Lu, Q. Zhang, L. Lin, M. O’Laughlin, J. McMichael, K. Delehaunty, L. Fulton, V. Magrini, S. McGrath, R. Demeter, T. Vickery, J. Hundal, L. Cook, G. Swift, J. Reed, P. Alldredge, T. Wylie, J. Walker, M. Watson, S. Heath, W. Shannon, N. Varghese, R. Nagarajan, J. Payton, J. Baty, S. Kulkarni, J. Klco, M. Tomasson, P. Westervelt, M. Walter, T. Graubert, J. DiPersio, L. Ding, E. Mardis, and R. Wilson, "The Origin and Evolution of Mutations in Acute Myeloid Leukemia", Cell, vol. 150, pp. 264-278, 2012. http://dx.doi.org/10.1016/j.cell.2012.06.023
- R. Chowdhury, K.K. Yeoh, Y. Tian, L. Hillringhaus, E.A. Bagg, N.R. Rose, I.K.H. Leung, X.S. Li, E.C.Y. Woon, M. Yang, M.A. McDonough, O.N. King, I.J. Clifton, R.J. Klose, T.D.W. Claridge, P.J. Ratcliffe, C.J. Schofield, and A. Kawamura, "The oncometabolite 2‐hydroxyglutarate inhibits histone lysine demethylases", EMBO reports, vol. 12, pp. 463-469, 2011. http://dx.doi.org/10.1038/embor.2011.43
- M. Toyota, N. Ahuja, M. Ohe-Toyota, J.G. Herman, S.B. Baylin, and J.P. Issa, "CpG island methylator phenotype in colorectal cancer.", Proceedings of the National Academy of Sciences of the United States of America, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10411935
- D.J. Weisenberger, K.D. Siegmund, M. Campan, J. Young, T.I. Long, M.A. Faasse, G.H. Kang, M. Widschwendter, D. Weener, D. Buchanan, H. Koh, L. Simms, M. Barker, B. Leggett, J. Levine, M. Kim, A.J. French, S.N. Thibodeau, J. Jass, R. Haile, and P.W. Laird, "CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer", Nature Genetics, vol. 38, pp. 787-793, 2006. http://dx.doi.org/10.1038/ng1834
- S.C. Mack, H. Witt, R.M. Piro, L. Gu, S. Zuyderduyn, A.M. Stütz, X. Wang, M. Gallo, L. Garzia, K. Zayne, X. Zhang, V. Ramaswamy, N. Jäger, D.T.W. Jones, M. Sill, T.J. Pugh, M. Ryzhova, K.M. Wani, D.J.H. Shih, R. Head, M. Remke, S.D. Bailey, T. Zichner, C.C. Faria, M. Barszczyk, S. Stark, H. Seker-Cin, S. Hutter, P. Johann, S. Bender, V. Hovestadt, T. Tzaridis, A.M. Dubuc, P.A. Northcott, J. Peacock, K.C. Bertrand, S. Agnihotri, F.M.G. Cavalli, I. Clarke, K. Nethery-Brokx, C.L. Creasy, S.K. Verma, J. Koster, X. Wu, Y. Yao, T. Milde, P. Sin-Chan, J. Zuccaro, L. Lau, S. Pereira, P. Castelo-Branco, M. Hirst, M.A. Marra, S.S. Roberts, D. Fults, L. Massimi, Y.J. Cho, T. Van Meter, W. Grajkowska, B. Lach, A.E. Kulozik, A. von Deimling, O. Witt, S.W. Scherer, X. Fan, K.M. Muraszko, M. Kool, S.L. Pomeroy, N. Gupta, J. Phillips, A. Huang, U. Tabori, C. Hawkins, D. Malkin, P.N. Kongkham, W.A. Weiss, N. Jabado, J.T. Rutka, E. Bouffet, J.O. Korbel, M. Lupien, K.D. Aldape, G.D. Bader, R. Eils, P. Lichter, P.B. Dirks, S.M. Pfister, A. Korshunov, and M.D. Taylor, "Epigenomic alterations define lethal CIMP-positive ependymomas of infancy", Nature, vol. 506, pp. 445-450, 2014. http://dx.doi.org/10.1038/nature13108
- A.D. Kelly, H. Kroeger, J. Yamazaki, R. Taby, F. Neumann, S. Yu, J.T. Lee, B. Patel, Y. Li, R. He, S. Liang, Y. Lu, M. Cesaroni, S.A. Pierce, S.M. Kornblau, C.E. Bueso-Ramos, F. Ravandi, H.M. Kantarjian, J. Jelinek, and J. Issa, "A CpG island methylator phenotype in acute myeloid leukemia independent of IDH mutations and associated with a favorable outcome", Leukemia, vol. 31, pp. 2011-2019, 2017. http://dx.doi.org/10.1038/leu.2017.12
- M. Kulis, S. Heath, M. Bibikova, A.C. Queirós, A. Navarro, G. Clot, A. Martínez-Trillos, G. Castellano, I. Brun-Heath, M. Pinyol, S. Barberán-Soler, P. Papasaikas, P. Jares, S. Beà, D. Rico, S. Ecker, M. Rubio, R. Royo, V. Ho, B. Klotzle, L. Hernández, L. Conde, M. López-Guerra, D. Colomer, N. Villamor, M. Aymerich, M. Rozman, M. Bayes, M. Gut, J.L. Gelpí, M. Orozco, J. Fan, V. Quesada, X.S. Puente, D.G. Pisano, A. Valencia, A. López-Guillermo, I. Gut, C. López-Otín, E. Campo, and J.I. Martín-Subero, "Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia", Nature Genetics, vol. 44, pp. 1236-1242, 2012. http://dx.doi.org/10.1038/ng.2443
- M. Kulis, A. Merkel, S. Heath, A.C. Queirós, R.P. Schuyler, G. Castellano, R. Beekman, E. Raineri, A. Esteve, G. Clot, N. Verdaguer-Dot, M. Duran-Ferrer, N. Russiñol, R. Vilarrasa-Blasi, S. Ecker, V. Pancaldi, D. Rico, L. Agueda, J. Blanc, D. Richardson, L. Clarke, A. Datta, M. Pascual, X. Agirre, F. Prosper, D. Alignani, B. Paiva, G. Caron, T. Fest, M.O. Muench, M.E. Fomin, S. Lee, J.L. Wiemels, A. Valencia, M. Gut, P. Flicek, H.G. Stunnenberg, R. Siebert, R. Küppers, I.G. Gut, E. Campo, and J.I. Martín-Subero, "Whole-genome fingerprint of the DNA methylome during human B cell differentiation", Nature Genetics, vol. 47, pp. 746-756, 2015. http://dx.doi.org/10.1038/ng.3291
- C.C. Oakes, M. Seifert, Y. Assenov, L. Gu, M. Przekopowitz, A.S. Ruppert, Q. Wang, C.D. Imbusch, A. Serva, S.D. Koser, D. Brocks, D.B. Lipka, O. Bogatyrova, D. Weichenhan, B. Brors, L. Rassenti, T.J. Kipps, D. Mertens, M. Zapatka, P. Lichter, H. Döhner, R. Küppers, T. Zenz, S. Stilgenbauer, J.C. Byrd, and C. Plass, "DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia", Nature Genetics, vol. 48, pp. 253-264, 2016. http://dx.doi.org/10.1038/ng.3488
- D.H. Spencer, D.A. Russler-Germain, S. Ketkar, N.M. Helton, T.L. Lamprecht, R.S. Fulton, C.C. Fronick, M. O’Laughlin, S.E. Heath, M. Shinawi, P. Westervelt, J.E. Payton, L.D. Wartman, J.S. Welch, R.K. Wilson, M.J. Walter, D.C. Link, J.F. DiPersio, and T.J. Ley, "CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression", Cell, vol. 168, pp. 801-816.e13, 2017. http://dx.doi.org/10.1016/j.cell.2017.01.021
- F. Tuorto, R. Liebers, T. Musch, M. Schaefer, S. Hofmann, S. Kellner, M. Frye, M. Helm, G. Stoecklin, and F. Lyko, "RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis", Nature Structural & Molecular Biology, vol. 19, pp. 900-905, 2012. http://dx.doi.org/10.1038/nsmb.2357
- S. Geyh, M. Rodríguez-Paredes, P. Jäger, C. Khandanpour, R. Cadeddu, J. Gutekunst, C.M. Wilk, R. Fenk, C. Zilkens, D. Hermsen, U. Germing, G. Kobbe, F. Lyko, R. Haas, and T. Schroeder, "Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia", Leukemia, vol. 30, pp. 683-691, 2015. http://dx.doi.org/10.1038/leu.2015.325
- J. Maksimovic, L. Gordon, and A. Oshlack, "SWAN: Subset-quantile Within Array Normalization for Illumina Infinium HumanMethylation450 BeadChips", Genome Biology, vol. 13, 2012. http://dx.doi.org/10.1186/gb-2012-13-6-r44
- Y. Chen, M. Lemire, S. Choufani, D.T. Butcher, D. Grafodatskaya, B.W. Zanke, S. Gallinger, T.J. Hudson, and R. Weksberg, "Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray", Epigenetics, vol. 8, pp. 203-209, 2013. http://dx.doi.org/10.4161/epi.23470
- J. MacDonald, "Affycoretools: Functions useful for those doing repetitive analyses with Affymetrix GeneChips.", R package version 1.46.5., 2008.
- G. Warners, B. Bolker, L. Bonebakker, R. Gentleman, W. Huber, A. Liaw, T. Lumley, M. Maechler, A. Magnusson, S. Moeller, M. Schwartz, and B. Venables, "gplots: Various R Programming Tools for Plotting Data.", R package version 3.0.1., 2016.
- E. Neuwirth, "ColorBrewer Palettes.", R package RColorBrewer version 1.1-2., 2014.
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
We thank the DKFZ Genomics and Proteomics Core Facility for Infinium EPIC methylation assay services, Jessica Eisel for technical and Felix Bormann for bioinformatical support. Stefan Pusch and Andreas von Deimling are patent holders of “Means and methods for the determination of (D)-2-hydroxyglutarate (D2HG)”, the enzymatic 2-HG assay used for D-2-HG determination in this manuscript (WO2013127997A1). The patent is under the administrative supervision of the DKFZ technology transfer office. All other authors declare no conflict of interests. This work was supported by a grant from Deutsche Forschungsgemeinschaft to F.L. (SPP1463).
COPYRIGHT
© 2017

mIDH-associated DNA hypermethylation in acute myeloid leukemia reflects differentiation blockage rather than inhibition of TET-mediated demethylation by Wiehle et al. is licensed under a Creative Commons Attribution 4.0 International License.


