Reviews:
Cell Stress, Vol. 5, No. 5, pp. 55 - 72; doi: 10.15698/cst2021.05.248
Cellular mechanisms linking cancers to obesity

1 Department of Biomedicine, University of Bergen, N-5020 Bergen, Norway.
# Equal contribution.
Keywords: obesity, cancer, mouse models, adipokines, metabolism, inflammation, extracellular Matrix remodeling.
Abbreviatons:
ARG2 – arginase 2;
BMI – body mass index;
COX2 – cyclooxygenase-2;
DIO – diet-induced obesity;
ECM – extracellular matrix;
FABP – fatty acid binding protein;
FACS – fluorescence-activated cell sorting;
FFA – free fatty acid;
HCC – hepatocellular carcinoma;
HFD – high-fat diet;
IGF – insuline growth factor;
IL – interleukin;
LFD – low-fat diet;
MMTV – mouse mammary tumor virus;
NASH – non-alcoholic steatohepatitis;
PDAC – pancreatic ductal adenocarcinoma;
PPARγ – peroxisome proliferator-activated receptor γ;
PyMT – polyoma middle T antigen;
S1P – sphingosine-1-phosphate;
SCFA – short-chain fatty acid;
TGFα – transforming growth factor-alpha;
TNFα – tumor necrosis factor-α.
Received originally: 27/01/2021 Received in revised form: 23/03/2021
Accepted: 26/03/2021
Published: 12/04/2021
Correspondence:
Nils Halberg, Department of Biomedicine, University of Bergen, Jonas Lies vei 91, 5020 Bergen, Norway; Phone: +47 5558 6442; nils.halberg@uib.no
Conflict of interest statement: The authors declare no competing interests.
Please cite this article as: Xiao-Zheng Liu, Line Pedersen and Nils Halberg (2021). Cellular mechanisms linking cancers to obesity. Cell Stress 5(5): 55-72. doi: 10.15698/cst2021.05.248
Abstract
Obesity is epidemiologically linked to 13 forms of cancer. The local and systemic obese environment is complex and likely affect tumors through multiple avenues. This includes modulation of cancer cell phenotypes and the composition of the tumor microenvironment. A molecular understanding of how obesity links to cancer holds promise for identifying candidate genes for targeted therapy for obese cancer patient. Herein, we review both the cell-autonomous and non-cell-autonomous mechanisms linking obesity and cancer as well as provide an overview of the mouse model systems applied to study this.
INTRODUCTION
Obesity is defined as an excessive accumulation of adipose tissue, which stores energy in the form of triglycerides. This mainly occurs when caloric intake exceeds energy expenditure. Currently, 1.9 billion adults and 38 million children worldwide are categorized as overweight – numbers that have tripled since 1975 [1]. Dating back to the mid-1980’s, clinicians reported that obese patients diagnosed with breast cancer have poorer survival outcomes [2]. Large epidemiological studies have since supported and expanded on these observations and estimates now suggest that obesity is implicated in 15-20% of cancer-related mortalities (reviewed in [3][4]). This is a consequence of obesity’s association with an increased risk of developing cancers (cancer incidence) and with specific obesity-linked biological effects on cancer progression in combination. Currently, obesity has been associated with 13 cancer types (reviewed in [5]). Epidemiological cross-comparisons have revealed that these interactions involve biological specificities. Examples include: i) sex-related correlations – the association between obesity and colon cancer are significantly more pronounced in males than in females, ii) specific histological subtypes – obesity is correlated with esophageal adenocarcinoma, but not esophageal squamous cell carcinoma and iii) menopause state – the risk of breast cancer is associated with obesity in postmenopause, but not in premenopause [6].
–
The obese environment is highly complex and comprises changes in serum levels of multiple circulating factors such as glucose, leptin, glucagon, adiponectin, glucagon-like-peptide-1, insulin, free fatty acids (FFA) and cholesterol; altered sleeping behavior; altered gut microbiome; altered pharmacodynamics of therapeutics, poor wound healing and post-operative infections, and the development of comorbid diseases (including chronic low-grade inflammation, heart disease, type 2 diabetes and hypertension). Such phenotypic complexity is present both systemically and in local tissue microenvironments. From the emerging molecular understanding of how such complexity in the obese environment links to cancers, we are beginning to uncover new and fascinating interactions between the physiological state of host and tumor behavior.
–
To recapitulate the complex obesogenic environment and uncover the molecular mechanisms that link obesity and cancer the field has so far relied on mouse models. In this review, we provide an overview of these model systems as well as proposed molecular and cellular processes by which the obese environment affects tumor initiation and progression (summarized in Figure 1).
–
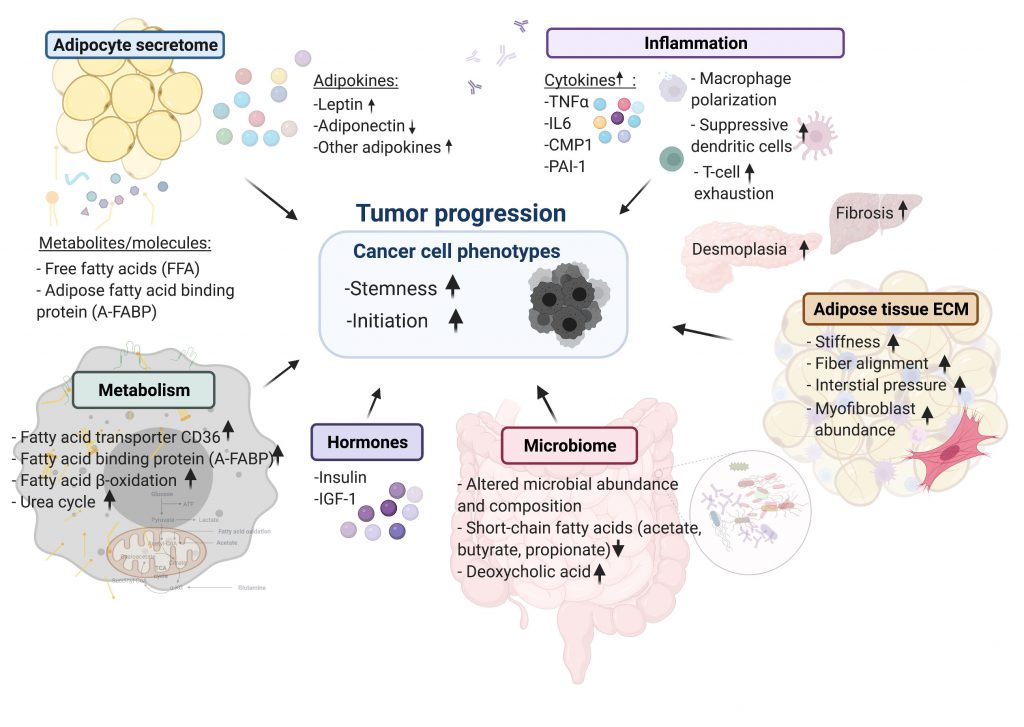 | FIGURE 1: Summary of cellular mechanisms linking cancers to obesity. |
ANIMAL MODELS OF THE OBESITY-CANCER CONNECTION
As in vitro models generally do not demonstrate the full complexity of the obesogenic phenotype, in vivo animal models have been essential research tools to address the mechanisms underlying the obesity-cancer connection. The most widely used models are either genetic or diet-induced obesity (DIO) models. Upon a cancer challenge these models broadly display increased tumor initiation and growth, relative to their non-obese counterparts. Importantly, these models often support the biological specificities of the epidemiological link between obesity and cancer. For example, consistent with human data, obesity leads to increased tumor burden and faster disease progression in oncogenic Kras-driven pancreatic ductal adenocarcinoma (PDAC), but not in lung cancer [7]. Table 1 presents an overview that details how both genetic and DIO models have been applied to cancer research.
–
–
Genetic animal models
Amongst genetic models, mice with deficient leptin signaling are the most frequently used [8]. In these models, obesity is caused by the lack of leptin (the ob/ob mouse), or by a mutation in the leptin receptor gene (db/db mouse), both causing the mice to overfeed. The genetic models consistently induce an early onset obese state and comorbidities such as insulin resistance and hepatic steatosis when the mice are fed standard chow. Their primary disadvantage is the exclusion of potential peripheral effects of leptin on cancer cells and the cells of the tumor microenvironment.
–
DIO animal models
The DIO rodent models are established by feeding animals with diets containing a high proportion of fat, sugar or a combination of the two. While several feeding patterns have been developed, the most commonly-used diets contain 30-60% kcals from fat which is provided to the mice for ten to twelve weeks prior to cancer challenge (Table 1). The long-term establishment period typically mimics the gradual development of obesity in humans as well as obesity-induced comorbidities that follows human obesity. On the other hand, this model is confounded by the potential biases created by the chosen dietary composition [9]. This could affect tumor outcomes independently of the obese phenotype per se and has led many investigators to include both DIO and genetic models [10][11][12][13]. While findings from the two independent model systems generally overlap, opposing results have been reported for intestinal cancer. In this study the authors reported that DIO induced intestinal tumor stemness, the db/db system led to a reduction in intestinal stem cell counts [12].
–
Given that the majority of comorbidities seen in obese individuals are dependent on obesity-induced inflammation [14], the field has relied on syngeneic transplants rather than xenograft models. Major challenges associated with syngeneic models include limited tumor heterogeneity, variable phenotypes depending on injection site, and relatively few transplantable cell lines as exemplified by an extreme reliance on E0771, a murine mammary carcinoma cell line and originally isolated as a spontaneous tumor from C57BL/6 mouse [15] (Table 1). Finally, the clinical specificities in the links between obesity and cancer create additional challenges in the model setup. Perhaps the best example of such is the dichotomy between obesity-induced breast cancer in pre- and postmenopausal women. Ovariectomy is a well-established method to induce post-menopausal state in mice [16]. After removal of the ovary, endogenous estrogen production stops and the ovariectomized mice usually develop increased body weight, hepatic steatosis, insulin resistance and impaired glucose tolerance [17]. Subcutaneously implanted tumors grow faster in the ovariectomized model, regardless of diet, and this makes it difficult to distinguish the effects of high-fat diet (HFD) on tumor progression from the effects of postmenopausal loss of hormones [18].
–
To establish HFD-induced obese mouse models, the feeding period varies from four to 24 weeks, although ten to twelve weeks is commonly used (Table 1). This period is sufficient to induce obese phenotypes featuring increased body weight, elevated fasting glucose levels, hyperinsulinemia, insulin resistance, hepatic steatosis, dyslipidemia and hypertension [19]. Such long-term HFD feeding is fairly consistently associated with enhanced tumor initiation and growth. In contrast, shorter HFD feeding periods of just four days is not associated with changes in body weight or features of metabolic syndrome. Such short-term feeding regiments do not affect tumor initiation of orthotopically implanted mouse breast cancer cells [20]. This was evident even though the HFD exposure was maintained for seven weeks until the end of the experiment [20]. Thus, an established state of obesity and obesity-induced physiological changes are required for affecting tumor outcomes. Short feeding periods should thus be avoided [20][21], and the establishment of obesity-induced clinical and biochemical features should be verified before tumor challenge.
–
Reversibility of obesity-induced effects on tumor outcomes
A key question in the field is to what degree reversing the obese phenotype affects tumor outcomes. In the ob/ob model, reestablishing leptin expression through adenoviral expression in muscle reversed the obese phenotype and related biochemical parameters, leading to the suppression of pancreatic cancer progression [7]. In the same study, the enhanced tumor progression in the ob/ob model was also reversed by dietary caloric restriction. With the DIO models, a number of studies have utilized dietary switch experiments. Five weeks low-fat diet (LFD) feeding after established DIO was enough to reverse obesity-induced inflammation and breast tumor formation [20]. A comparison between a short-term and a long-term dietary switch demonstrated that exposing the DIO mice to LFD for seven days was not sufficient to affect the obesity-induced effects on intestinal stem cells, while the phenotype was reversed after four weeks LFD [12]. Taken together, these observations indicate that obesity-induced effects on tumor outcomes are reversible.
MOLECULAR MECHANISMS OF THE OBESITY-CANCER CONNECTION
The interaction between obesity and cancer could be governed by genetic as well as non-genetic alterations. Currently there is limited evidence for a genetic component linking obesity to cancer initiation. This likely reflects the difficulty of obtaining body mass index (BMI) measures as well as incorporation of potential cachexia-induced weight loss in large consortia databanks containing patient genomic status. Instead, an expanding number of cellular processes have been suggested to govern obesity effects in malignancies.
–
Adipokines affecting carcinogenesis
Adipose tissue is an active endocrine organ which secretes adipokines, chemokines and constituents of the extracellular matrix (ECM) [22]. In response to increased demand of triglyceride storage, adipose tissue undergoes hyperplasia and hypertrophy in the transition from a lean to an obese state. These architectural adjustments induce changes in the adipocyte secretome and released metabolites which contribute to an altered micro- and macroenvironment in the obese compared to the lean setting [22].
–
Leptin
–
Adipokines have long been hypothesized to provide a mechanistic link between cancer and obesity. Leptin is secreted in proportion to adipose volume and total fat mass [23][24]. By signaling through leptin receptors in the central nervous system (CNS), leptin regulates energy balance by functioning as a satiety signal, which reduces food intake and increases energy expenditure. In obese individuals, however, the satiety-promoting effects of leptin are impaired by the induction of cellular leptin resistance [25]. Along with decreasing energy expenditure and promoting obesity, hyperleptinemia has pronounced peripheral effects on cancer cells (discussed below) and the tumor microenvironment including immune cells – particularly the T-helper 1 cells [26][27].
–
Leptin and the leptin receptors have been identified in malignant cells of various origins including hepatocellular cancer, colorectal cancer, thyroid cancer and breast cancer [28]. Overall, leptin has been assigned a pro-tumorigenic function. In breast cancer, leptin and the leptin receptor are overexpressed in primary and metastatic lesions compared to non-cancer tissues [29][30]. In cell culture, leptin acts as a growth-stimulating agent for breast cancer cells, promoting proliferation and repressing apoptotic pathways [31]. A similar role has been reported in colon cancer, where leptin acts as a growth factor at stages correlating with tumor initiation in a murine model [32]. Moreover, leptin treatment of cancer cells modulates processes such as metabolic reprogramming [33] and reactive oxygen species production [34][35].
–
In females, higher leptin concentrations are associated with increased risk, as well as grade, stage and recurrence of breast cancer [36][37]. In the obese context, leptin has been linked to tumor-initiating cell survival and obesity-associated triple negative breast cancer development by promoting cancer stem cell enrichment and epithelial-to-mesenchymal transition (EMT) [38][39]. Leptin governs the stem cell phenotype through epigenetic mechanisms controlled by a leptin-STAT3-G9a histone methyltransferase signaling axis [40].
–
In sum, several studies support a role of leptin in tumorigenesis which favors cell growth and survival by increasing proliferation and decreasing apoptosis, regulating inflammatory processes, modulating cancer stem cell properties as well as metabolic activity. However, augmented tumor formation is observed for several cancers, including cancers of the liver, kidney and pancreas in ob/ob and db/db compared with wildtype mice [7][11][41][42] suggesting leptin-independent mechanisms. A number of the early mammary cancer studies were performed with the mouse mammary tumor virus (MMTV)-transforming growth factor-alpha (TGF-α) transgenic mouse model – a cancer model in which TGF-α overexpression is under the control of the MMTV promoter. Without any further genetic alteration, 30-40% of MMTV-TGF-α mice develop spontaneous tumors within 16 months [43]. However, the overexpression of TGF-α in the background of either ob/ob or db/db failed to develop tumors, suggesting a required role of leptin signaling in TGF-α driven carcinogenesis [44][45]. Interestingly, MMTV-Wnt1 cell-derived syngeneic breast cancer cells displayed increased mammary tumor burden upon implantation into db/db mice, whereas implantation of the same cells into ob/ob mice did not affect tumor growth [38]. This suggests that peripheral cancer cell leptin signaling is required for obesity-dependent effects of MMTV-Wnt1 cells. Taken together, these experiments illustrate that leptin does not solely account for obesity-accelerated tumor progression for all cancer types.
–
Adiponectin
–
In addition to leptin, deregulation of the adipokine adiponectin has been implicated in driving tumor progression in several obesity-associated cancer types, including colon, liver, breast, renal, gastric, esophageal, pancreatic and endometrial cancer [46]. Circulating adiponectin levels negatively correlate with body fat mass and adiposity. This hormone acts as an insulin sensitizer of tissues such as liver and muscle, as well as balancing glucose and lipid metabolism [47][48]. Adiponectin mediates its effects through its classical and ubiquitously expressed receptors AdipoR1 and AdipoR2, and the non-classical receptor T-Cadherin [49][50]. Several signaling pathways are stimulated by adiponectin, including AMPK, PI3K/AKT, MAPK, mTOR, NF-kB and STAT3 [46]. Adiponectin exerts both anti-inflammatory and anti-proliferative effects and is hence often referred to as the “guardian angel adipokine” [48][51]. In addition, adiponectin also potently stimulates ceramidase activity through AdipoR1 and AdipoR2, and thereby enhance pro-apoptotic ceramide catabolism leading to formation of its downstream anti-apoptotic metabolite sphingosine-1-phosphate (S1P) [52]. HFD is sufficient to induce expression of the enzyme that generates S1P (sphingosine kinase 1, SphK1) and its receptor S1P receptor 1 (S1PR1) in syngeneic and spontaneous breast tumors [53]. Targeting the SphK1/S1P/S1PR1 axis in these models attenuates obesity-induced tumor progression [53].
–
Epidemiological studies have linked low serum adiponectin levels to an increased risk of colon cancer [54]. This is consistent with in vivo studies that have demonstrated promotion of intestinal carcinogenesis by lack of adiponectin in both genetic and chemically induced cancer models [55]. Another study also found a repressing role on colonic epithelial proliferation in a chemically induced cancer model. However, this effect was specific to adiponectin-deficient mice fed an HFD. No effect was observed for adiponectin-deficient mice fed a basal diet compared with wild-type mice [56]. Likewise, adiponectin protects against liver tumorigenesis in nude mice, and its reduced expression is associated with poor prognosis in obese patients with hepatocellular carcinoma [57]. These findings were supported by studies using an orthotopic liver tumor nude mouse model, which showed inhibited tumor growth and lower incidence of lung metastasis in adiponectin treated mice [58]. Furthermore, adiponectin treatment suppressed hepatic stellate cell activation and macrophage infiltration [58]. In addition to anti-inflammatory and growth-suppressing effects, adiponectin has been shown to constrain tumor growth by inhibiting tumor vasculature [58][59]. On the contrary, lack of adiponectin in a MMTV-polyoma middle T antigen (PyMT) model significantly reduced tumor growth and angiogenesis [60][61]. Thus, a general role of adiponectin in tumor angiogenesis remains to be defined.
–
Other adipokines
–
In addition to altered levels of leptin and adiponectin, increased levels of obesity-related adipokines such as interleukin-6 (IL-6), interleukin-8 (IL-8), tumor necrosis factor-α (TNF-α), visfatin and resistin are associated with increased cancer risk [62]. Resistin is mainly expressed in adipocytes and immune cells [63] and has been reported to promote breast tumorigenesis in vitro and in vivo[64]. In a DIO mouse model, its expression and secretion are significantly increased in mammary adipose tissue. Mechanistically, HFD-induced elevated levels of circulating FFA promote peroxisome proliferator-activated receptor γ (PPARγ) signalling, which is an upstream regulator of WW-domain-containing transcription regulator 1 (WWTR1; also known as TAZ) expression. The enhanced TAZ expression via the FFA/PPARγ axis further activates resistin secretion [64].
–
Adipose tissue is a major component of the breast. The cytokine secretion from adipocytes is affected by menopausal status, which could link to the paradoxical association of obesity and pre- and post-menopausal breast cancer risk. After menopause, a high estrone (E1): estradiol (E2) ratio in tissue and circulation was observed in both mouse models and human patients. A high intratumor E1:E2 ratio cooperates with NFκB pathway to increase tumour stemness in obesity [65].
–
In summary, accumulating evidence obtained by a variation of model systems supports an important role of adipokines in tumor progression. On the other hand, knowledge derived from a curious counterpart to the obese phenotype, the fatless A-Zip/F1 mouse, challenges a universal role. The fatless mouse model mimics generalized lipodystrophy and is characterized by the majority of the comorbidities associated with obesity including insulin resistance and ectopic lipid accumulation but lack the secretion of adipokines. In chemical carcinogenesis and transgenic C3(1)/T-Ag mammary models, tumor incidence was consistently higher in the fatless mouse compared with controls – suggesting that adipocyte-independent factors drive tumorigenesis in this environment [66]. Overall, more model-specific research is needed to fully understand the link between obesity, cancer and adipokines.
–
Insulin and insulin-like growth factor-1 (IGF-1)
Increased serum concentrations of insulin and IGF-1 are frequently detected in obese individuals. Both factors have been proposed to serve as key hormonal mediators mechanistically linking obesity and cancer [67]. Insulin and IGF-1 are closely related ligands that activate the insulin receptor (INSR) and IGF-1 receptor (IGF-1R), leading to stimulation of two key cell proliferation and protein synthesis pathways for tumorigenesis – the PI3K-AKT-mTOR signaling pathway and the RAS-MAPK pathway (reviewed in [68]). Expression of the respective receptors is detected on cancer cells of different origins and several cancers are driven by insulin and IGF-1 in vitro[69][70][71][72][73]. A positive association is also detected between insulin and obesity-prone cancers in terms of tumor growth in rodents as well as stage at diagnosis and death in humans (reviewed in [74]). Moreover, case-control and cohort studies have demonstrated that individuals with higher levels of insulin or C-peptide (indirect insulin measure) are at higher risk for developing obesity-related cancers including breast, endometrial, colorectal, pancreatic, liver, ovarian and gastric cancers compared to individuals with low levels of these factors (reviewed in [75]).
–
In addition to a mitogenic function in carcinogenesis, insulin has been suggested to effect metabolic processes in cancer cells. Insulin increases mitochondrial glucose oxidation and augments cell division in cells derived from obesity-associated tumors, including colon, breast and prostate cancer. In contrast, no alteration of substrate preference is observed in obesity-independent cell lines (melanoma, lymphoma and small cell lung cancer) [76]. In studies using an insulin lowering agent, dapagliflozin, a sodium-glucose cotransporter-2 (SGLT2) inhibitor, reduction in insulin levels slowed obesity-accelerated tumor growth of both syngeneic breast and colon cancer models. The authors concluded that this effect is not due to increases in ketosis or to a direct effect on tumor cell division, but rather is mediated by the reversal of hyperinsulinemia, resulting in diminished tumor glucose uptake and oxidation [77]. At present, several human clinical trials are investigating insulin-lowering treatments as adjuvants to cancer treatments (reviewed in [74]).
–
ECM remodeling and fibrosis
Tumors are composed of cancer cells along with their connected stromal compartment. The tumor stroma consists of vasculature, non-transformed cell types including fibroblasts and immune cells, as well as structural elements such as the basement membrane and ECM. The stromal compartment is an integral part of cancer initiation, growth and progression (reviewed in [78]) and a number of studies have begun to delineate how obesity-induced stromal alterations influence tumor growth. Adipose tissue in obese individuals is among others characterized by an altered biochemical as well as biophysical microenvironment [79]. For instance, adipocytes in expanding adipose tissue deposit altered amounts of ECM components causing ECM remodeling and changes in tissue stiffness. Furthermore, obese tissue ECM contains more aligned fibers and higher interstitial pressure (reviewed in [80]). These changes are not restricted to subcutaneous and visceral fat but occur also in other fat depots. For instance, the homeostasis of mammary fat is disrupted in obese mouse models, as this tissue is enriched in myofibroblasts and stiffness-promoting ECM components [81]. These alterations, which are characteristic features of desmoplasia and fibrosis, can promote the tumorigenic potential of premalignant human breast epithelial cells [81]. Research on mouse models has shown that obesity also promotes desmoplasia in the pancreas, a common histological feature of PDAC, which is associated with accelerated tumor growth and impaired delivery/efficacy of chemotherapeutics through reduced perfusion [42]. Interestingly, it was suggested that the exacerbated desmoplasia and augmented tumor growth was a result of crosstalk between adipocytes, tumor-associated neutrophils and pancreatic stellate cells [42]. Grohmann et al. reported how obesity-associated hepatic stress could independently contribute to the pathogenesis of non-alcoholic steatohepatitis (NASH), fibrosis and hepatocellular carcinoma (HCC). Obesity promotes hepatic STAT1 dependent T-cell infiltration, NASH and fibrosis as well as NASH-independent STAT3-dependent HCC [82].
–
Moreover, single ECM components that are deregulated in the obese state have been demonstrated to promote tumorigenesis. One example is collagen VI, which is abundantly produced and secreted by adipocytes. A recent study applying proteomic analysis of ECM isolated by in vitro decellularization methods identified collagen VI to be up-regulated in murine obese mammary gland and breast tumor tissues relative to lean tissues [83]. Adipocyte-derived collagen VI has previously been demonstrated to be important for early mammary tumor progression, as collagen knockout mice in the background of the MMTV-PyMT mammary cancer model have reduced rates of early hyperplasia and primary tumor growth [84]. In vitro studies point to that full-length collagen VI is a driver of triple negative breast cancer cell adhesion, migration and invasion [83].
–
Furthermore, obesity-associated ECM remodeling has been shown to regulate the properties of immune cells. Culturing of bone-marrow-derived macrophages on ECM isolated from obese mice induce proliferation, changes the morphology, impact polarization as well as promote angiogenic traits compared to cells cultured on top of ECM derived from lean mice [85]. Hermano et al. reported that macrophage function could be regulated by heparanase, an endoglucuronidase that cleaves heparin sulfate in ECM [86]. They provided evidence that heparanase preferently is expressed in clinical/experimental obesity-associated breast tumors, and that heparanase deficiency abolishes obesity-accelerated orthotopic tumor growth. In the obese environment, they showed that heparanase stimulates macrophage production of inflammatory mediators that induce aromatase, a rate-limiting enzyme in estrogen synthesis [86].
–
Cancer metabolism
Metabolic reprogramming is commonly observed in cancer cells during tumor progression. Given the multiple local and systemic metabolic abnormalities in obese individuals, the obese state provides an interesting case for new discoveries in the interface between systemic and tumor metabolism.
–
The effects of FFA on tumor initiation
–
Recent studies have proposed that lipid metabolism and fatty acid oxidation contribute to cancer stemness. A subpopulation of human carcinoma cells that expressed high levels of the fatty acid transporter CD36 and lipid metabolism genes was identified to possess cancer stemness features and to contribute to a poor prognosis in human cancer patients [87][88]. In intestinal cancers, exposure to FFA was demonstrated to activate PPARδ-dependent signaling to endow tumor-initiating capacity [12]. In addition to direct cancer cell-autonomous effects, obesity-induced elevated levels of saturated fatty acid drive the accumulation of metabolically activated macrophages in adipose tissue. Such macrophages were shown to enhance stem-like properties in triple negative breast cancer cells and promote tumor initiation in obese mouse models [20]. Overall, there is compelling evidence that fatty acids could be a critical link between obesity and tumor initiation.
–
The effects of FFA on tumor progression
–
In addition to enhanced tumor initiation, emerging evidence indicates that the elevated circulating FFA and fatty acid binding proteins in the obese state are associated with cancer progression. In ovarian cancer, the rapid growth and metastatic colonization of cancer cells were suggested to be directly fueled by fatty acids delivered by fatty acid binding protein 4 (FABP4), also known as adipocyte FABP (A-FABP), from adipocytes [89]. Furthermore, lipolysis in adipocytes and increased β-oxidation rate in cancer cells were observed in co-culture experiments, suggesting that adipocyte-derived fatty acids can act as an energy source fueling cancer cells [89]. Hao et al. expanded the role of FABP itself. They demonstrated that increased circulating levels of A-FABP enhanced breast cancer stemness and aggressiveness in both in vitro and in vivo models [90]. Moreover, the authors further determined that the association is dependent on STAT3/ALDH1 signaling regardless of endogenous A-FABP level, suggesting a dual mechanism by which A-FABP supports tumor progression both as a fatty acid transporter and as a signaling molecule [90]. Consistently, Madak-Erdogan et al. demonstrated that obesity-induced elevated circulating FFA could be taken up by estrogen-positive breast cancer cells and promote cancer proliferation in obese postmenopausal women [91]. Also, these findings suggested that fatty acids activate a mTOR and MAPK dependent signaling network to facilitate increased glycolytic and aerobic respiration in breast cancer cells. Finally, cancer cells and immune cells display distinct metabolic adaptations in the obesogenic tumor microenvironment [92]. For example, HFD represses PHD3 expression specifically in cancer cells, which rewire their metabolism to accelerate fatty acid uptake and oxidation. This in turn impacts the fatty acid availability for T cells in the same microenvironment and thereby impairs T cells infiltration and function [92].
–
Other metabolites and tumor progression
–
In addition to fatty acid-related adaptations, modifications in other metabolic pathways have been identified in tumors evolving in obese environments, including glucose handling, nitrogen metabolism and pyruvate-dependent mitochondrial respiration. For example, in a genetic model for pancreas cancer, HFD feeding heightened aerobic glycolysis through hyperactivation of oncogenic KRAS [93]. Moreover, transcriptomic analysis of an orthotopic xenograft PDAC model revealed an enrichment of nitrogen metabolism pathways [94]. The authors identified the mitochondrial enzyme arginase 2 (ARG2), which catabolizes arginine into ornithine and urea, to be induced in obese mouse tumors and that its expression level correlated with patient BMI. As an important enzyme at the final step of the urea cycle, the obesity-induced upregulated ARG2 enhances nitrogen flux into the urea cycle. Moreover, the depletion of ARG2 causes ammonia accumulation and suppresses PDAC, particularly in obese hosts [94]. In a search for mechanisms underlying the increased cancer risk that is associated with the combination of metabolic deregulation and circadian disruption, Ramos et al. demonstrated that a non-canonical function of BMAL1 limits obesity-promoted triple-negative breast cancer [95]. BMAL1, a key circadian transcription factor, suppresses the flexibility of mitochondrial substrate usage and pyruvate-dependent mitochondrial respiration induced by chronic insulin treatment in vitro. Interestingly, orthotopic transplantation of E0771 breast cancer cells depleted for BMAL1 revealed that BMAL1 functions as a tumor suppressor in obese, but not in lean mice. In humans, down-regulation of BMAL1 is associated with higher risk of metastasis [95].
–
Inflammation
Obesity is associated with local and systemic immune system dysregulation characterized by increased secretion of inflammatory cytokines and phenotypic conversions in immune cells [96]. Recently, a growing number of studies have suggested that obesity-associated inflammatory alterations might promote progression of multiple cancer types through diverse mechanisms.
–
Tumor immune cell abundances
–
A fundamental question is whether obesity-induced chronic inflammation alters immune cell infiltration in tumors. At the histological level, HFD-feeding increases infiltration of tumor-associated macrophages in both transplant and some autochthonous PDAC models [97][98][99] as well as in transplant models of breast cancer [53][86] and in chemically induced hepatocarcinoma [100][101]. In contrast, transcriptional analysis of the duodenum of K-rasG12Dint mice revealed down regulated F4/80 expression in mice fed HFD [102], and fluorescence-activated cell sorting (FACS) analysis of transplanted E0771 breast tumors showed a reduction of CD11b+F4/80+ macrophages [103]. In kidney cancer, increased numbers of inhibitory dendritic cells (DC) infiltrate tumors of DIO mice [104]. Similar observations have been reported in breast cancer, as a population of CD11b+F4/80− cells (consisting of neutrophils and monocytes) identified by FACS showed a 31% increase in E0771 tumors from HFD mice [103]. Taken together, conflicting results on obesity-induced immune cell tumor infiltration has been reported for different cancer types as well as for different cancer/obesity models applied. This is in line with our own preliminary findings. We interrogated the immune-infiltrating cells in tumors grown in obese and non-obese mouse models using a 36-marker immune-focused mass cytometry panel [105]. Immunotyping of three syngeneic breast cancer models and two pancreas cancer models revealed that tumor immune infiltrate composition is highly model- and cancer type-specific. While no major immune cell alterations were observed in the pancreas cancer models and in two of the breast cancer models, HFD-feeding increased two T cell suppressive cell types and decreased CD8 T-cells in the E0771 breast cancer model [105].
–
Macrophages
–
In addition to affecting the overall infiltration of immune cells, accumulating evidence indicates that obesity affects immune function through modulating immune cell phenotypes. The immune subset that has so far received the most attention is macrophages. In several cancer models, obesity has been proposed to regulate recruitment, polarization and signaling of macrophages, thereby contributing to accelerated tumor progression. In breast and pancreatic cancer models, obesity induces recruitment of tumor-associated macrophage (TAM) with an M2-like cytokine profile. VEGFR-1 is abundantly expressed in TAMs, and blockade of VEGFR-1 signaling in obese but not lean mice leads to a shift in pro-tumor cytokine production and TAM polarization from an M2 pro-tumor to an M1 anti-tumor phenotype that ultimately reduce obesity-induced tumor progression [97]. Furthermore, the elevated level of fatty acids during obesity drives polarization of adipose tissue macrophages towards a metabolically activated phenotype. This phenotypic shift alters the niche to support triple negative breast cancer stemness and tumorigenesis through an IL-6/GP130 signaling axis [20]. In colitis-associated colorectal cancer, signaling through obesity-induced IL-6 was reported to shift macrophage polarization towards a tumor-promoting phenotype that produced the chemokine CC-chemokine-ligand-20 (CCL-20) in the tumor microenvironment. CCL-20 in turn, promoted tumorigenesis by recruiting CC-chemokine-receptor-6 (CCR-6)-expressing γδ T and B cells via chemotaxis [106]. Obesity-accelerated cancer growth is also associated with increased IL-6 levels and shift in M2/M1 macrophage ratio in a genetic mouse model of prostate cancer. Interestingly, these effects could specifically be blocked in HFD-fed mice by inhibition of the IL-6 receptor [107].
–
In two syngeneic orthotopic transplant models, a NLRC4 inflammasome/IL-1β signaling was demonstrated to provide a link between obesity and breast cancer progression. The obese tumor microenvironment induced an increase in the number of tumor-infiltrating macrophages with an activated NLRC4 inflammasome, which further activated IL-1β. The NLRC4/IL-1β module was able to promote angiogenesis via up-regulation of VEGFA, which in turn to contributed tumor progression [108]. In a liver cancer model, HFD induced liver endoplasmic reticulum (ER) stress boosted macrophages-mediated production of inflammatory cytokines. This in turn activated TNF receptor 1 (TNFR1)-IκB kinase β (IKKβ) signaling, which thereby contributed to hepatocyte proliferation and formation of HCC [101]. The importance of macrophages in the connection between obesity and breast cancer has however been challenged. Bousquenaud et al. reported that HFD tumors contained reduced amount of M1 macrophages, and that depletion of macrophages by clodronate liposomes boosted tumor growth in HFD mice. Hence, the authors concluded that macrophages do not contribute to promote obesity accelerated tumor progression [103].
–
T cells
–
Tumoral T cell function is of great interest, especially since the T cell is the critical mediator of immunotherapies, including adoptive T cell therapy and immune checkpoint therapy. Obese cancer patients have been reported to display increased levels of dysfunctional and exhausted T cells [92][109], suggesting that immunotherapy could be a promising treatment option for obese cancer patients. Indeed, in both human patients and preclinical animal models, obesity associates with better response to anti-PD1 and anti-CD8 immune checkpoint therapy [92][109][110].
–
Blocking obesity-induced inflammation
–
To address whether inflammatory processes are required for obesity-induced tumor initiation and progression, several studies have obstructed inflammatory signaling pathways, through genetic depletion of key cytokines or pharmacological inhibition of specific immune cell populations in animal models. Systemic knockout of TNF-α, IL-6 or TNFR1 all prevent obesity-induced cancer formation in HCC [100] and PDAC [99] suggesting a functional role of the immune apparatus. Although these studies are limited by the systemic depletion, they indicate a required role of the immune system in obesity-induced cancers.
–
Experimental evidence across cancer types supports a pivotal role of cyclooxygenase-2 (COX-2)-mediated inflammatory pathways in obesity promoted cancer progression [98][111]. A study applying an adult acinar-specific mutant Kras PDAC model, demonstrated that high fat consumption can activate and sustain Kras activity via COX-2 induction and COX-2 mediated positive feedback loops, which in turn contribute to the development of PanINs and PDAC. Inhibition and genetic knockout of COX-2 prevented pancreatic inflammation and initiation of PDAC, suggesting important roles for COX-2-dependent inflammatory processes [98]. In contrast, treatment with aspirin, a nonsteroidal anti-inflammatory agent (NSAID) that blocks the cyclooxygenase enzymes, did not impact tumor progression, immune cell infiltration or fibrosis in an obese genetic model of PDAC [7]. Consistently, the growth advantage of orthotopically implanted oncogenic Kras-driven PDAC cell lines in the obese environment was sustained in obese C57Bl6 mice lacking T-, B- and NK-cells (Rag2-/-::CD47-/-::Il2rg-/-) [105].
–
Microbiome
An integrated part of the obese phenotype is alterations in the microbiota composition [112][113]. As a natural part of the tumor microenvironment of gastrointestinal malignancies, the altered microbiota can directly participate in the regulation of gastrointestinal cancer progression. In addition to such local effects, microbiota-mediated metabolites display both systemic metabolic and inflammatory changes [114][115]. Gut microbiota are the major producers of short-chain fatty acids (SCFAs), which can be the utilized as an energy source and as key signaling molecules. Feeding mice with an HFD results in the decreased production of multiple SCFAs including acetate, propionate and butyrate and were demonstrated to promote G12D mutant Kras-driven intestinal carcinogenesis [102]. Interestingly, butyrate supplement through feeding successfully reversed the cancer progression effect of the high-fat feeding. Also, fecal-transplantation from HFD-fed to normal diet fed Kras mutant mice was sufficient to induce tumorigenesis [102]. These findings suggest that an HFD-induced microbiota shift synergizes with the Kras mutation during tumorigenesis and that such effects could act independently of obesity. In chemically-induced HCC, obesity-dependent alterations of the gut microbiota promote tumorigenesis through deoxycholic acid – a secondary bile acid [11][111]. This metabolite could cause DNA damage and provoke a senescence-associated secretory phenotype (SASP) in hepatic stellate cells to promote HCC development. In addition to bile acids, the altered gut microbiota provided lipoteichoic acid to the liver, and this compound synergistically promoted the SASP in hepatic stellate cells. Also, lipoteichoic acid is able to induce the COX-2-mediated immunosuppression in liver immune cells [111]. Altogether, the microbiota is an important component when studying gastrointestinal cancers in obesity. Due to the complex environment, further studies are required to understand and establish a solid mechanistic basis of the relationships between the microbiome, obesity and gastrointestinal cancers progression.
SUMMARY AND FUTURE PERSPECTIVES
As an expanding number of reports aiming to identify molecular connections between the obese state and tumors are released, it is clear that the complexity of the obese environment likely translates into integrated and complex molecular drivers. This necessitates the use of proper model systems that realistically recapitulates such environments. Whereas we have so far relied on mouse models, additional model systems as for example organoids grown in obese and non-obese conditions could become valuable alternatives to alleviate the limitations linked to mouse studies described above.
–
So far, the most consistent molecular link between the obesogenic environment and tumorigenesis revolves around increased abundance of fatty acids, ECM remodeling and immune cell differentiation. Fatty acids have been implicated in both tumor growth and tumor initiation through cell intrinsic and extrinsic mechanisms. In the tumor cells FFA have been suggested to act as an alternative fuel source while FFA have also been shown to modulate metabolically-activated macrophages and T-cells to create a pro-tumorigenic microenvironment. Direct effects of the obese environment on T-cells further translate into a potential added benefit of immune checkpoint therapy in the obese setting. Through tumor promoting modulation of the stromal compartment of the microenvironment, obesity leads to both biophysical changes driven by alterations in myofibroblast cell populations and biochemically through altered collagen VI secretion and processing.
–
So far, the vast majority of mechanistic studies have focused on obesity-dependent alterations in adipocytes, immune cells and their secreted molecules. However, recent work urges us to take a broader look at additional cell types that could be affected by the obese state. This, for example, include the islet cells of the pancreas. In the obese setting, these adapt by inducing Cck which in turn acts locally on pancreatic acinar to cells to drive tumorigenesis [7].
–
Another area of increasing interest is the connection between obesity and epigenetic deregulation. This connection was previously demonstrated in somatic tissues and has recently also been investigated in tumors [116]. Epigenetic rearrangements in cancers grown in obese mice, have been reported in prostate cancer [117], colorectal cancer [10] and breast cancer [118]. How the obese environment induces such changes is currently unknown, but it is clear that it could have profound effects on tumorigenesis. Interestingly, many metabolic intermediates including Acetyl-CoA and S-adenosyl methionine act as epigenetic precursors and can directly and indirectly affect chromatin modifications and function [119]. Going forward, it will be interesting to investigate if obesity-dependent changes in cellular metabolites are potential drivers of chromatin remodeling in tumors.
–
While the epidemiological link between obesity and cancer risk is well proven, the link between obesity and metastatic spread is less clear. This is reflected in far fewer mouse studies of metastasis in the obese setting. In one study, breast cancer lung metastatic colonization was enhanced in obese mice via GM-CSF- and IL-5 dependent recruitment of neutrophils [13]. Further studies in breast cancer using transplant and genetic models suggest that obesity-dependent changes in the sphingolipid profile is driving metastatic colonization in the obese setting [53]. However, potential mechanisms of obesity linked metastatic spread are currently understudied.
–
Finally, in addition to potentially revealing new exciting mechanistic pathways, discoveries of dependencies of the obesity-cancer link might lead to the development of novel targeted therapies for the benefit of growing group of obese cancer patients.
REFERENCES
- WHO (2020). Obesity and overweight. Available at: https://mdanderson.libanswers.com/faq/26219 [Last Accessed 07.03.2021]
- Greenberg ER, Vessey MP, McPherson K, Doll R, Yeates D (1985). Body size and survival in premenopausal breast cancer. Br J Cancer 51(5): 691-697. 10.1038/bjc.1985.104
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ (2003). Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348(17): 1625-1638. 10.1056/NEJMoa021423
- Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, Smeeth L (2014). Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5.24 million UK adults. Lancet 384(9945): 755-765. 10.1016/S0140-6736(14)60892-8
- Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K, International Agency for Research on Cancer Handbook Working G (2016). Body Fatness and Cancer–Viewpoint of the IARC Working Group. N Engl J Med 375(8): 794-798. 10.1056/NEJMsr1606602
- Renehan AG, Zwahlen M, Egger M (2015). Adiposity and cancer risk: new mechanistic insights from epidemiology. Nat Rev Cancer 15(8): 484-498. 10.1038/nrc3967
- Chung KM, Singh J, Lawres L, Dorans KJ, Garcia C, Burkhardt DB, Robbins R, Bhutkar A, Cardone R, Zhao X, Babic A, Vayrynen SA, Dias Costa A, Nowak JA, Chang DT, Dunne RF, Hezel AF, Koong AC, Wilhelm JJ, Bellin MD, Nylander V, Gloyn AL, McCarthy MI, Kibbey RG, Krishnaswamy S, Wolpin BM, Jacks T, Fuchs CS, Muzumdar MD (2020). Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma. Cell 181(4): 832-847 e818. 10.1016/j.cell.2020.03.062
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994). Positional cloning of the mouse obese gene and its human homologue. Nature 372(6505): 425-432. 10.1038/372425a0
- Speakman JR (2019). Use of high-fat diets to study rodent obesity as a model of human obesity. Int J Obes 43(8): 1491-1492. 10.1038/s41366-019-0363-7
- Li R, Grimm SA, Chrysovergis K, Kosak J, Wang X, Du Y, Burkholder A, Janardhan K, Mav D, Shah R, Eling TE, Wade PA (2014). Obesity, rather than diet, drives epigenomic alterations in colonic epithelium resembling cancer progression. Cell Metab 19(4): 702-711. 10.1016/j.cmet.2014.03.012
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499(7456): 97-101. 10.1038/nature12347
- Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ, Bauer-Rowe KE, Xifaras ME, Akkad A, Arias E, Pinello L, Katz Y, Shinagare S, Abu-Remaileh M, Mihaylova MM, Lamming DW, Dogum R, Guo G, Bell GW, Selig M, Nielsen GP, Gupta N, Ferrone CR, Deshpande V, Yuan GC, Orkin SH, Sabatini DM, Yilmaz OH (2016). High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531(7592): 53-58. 10.1038/nature17173
- Quail DF, Olson OC, Bhardwaj P, Walsh LA, Akkari L, Quick ML, Chen IC, Wendel N, Ben-Chetrit N, Walker J, Holt PR, Dannenberg AJ, Joyce JA (2017). Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol 19(8): 974-987. 10.1038/ncb3578
- O’Rourke RW (2009). Inflammation in obesity-related diseases. Surgery 145(3): 255-259. 10.1016/j.surg.2008.08.038
- Sugiura K, Stock CC (1952). Studies in a tumor spectrum. I. Comparison of the action of methylbis (2-chloroethyl)amine and 3-bis(2-chloroethyl)aminomethyl-4-methoxymethyl-5-hydroxy-6-methylpyridine on the growth of a variety of mouse and rat tumors. Cancer 5(2): 382-402. 10.1002/1097-0142(195203)5:2<382::aid-cncr2820050229>3.0.co;2-3
- Haslam SZ, Osuch JR, Raafat AM, Hofseth LJ (2002). Postmenopausal hormone replacement therapy: effects on normal mammary gland in humans and in a mouse postmenopausal model. J Mammary Gland Biol Neoplasia 7(1): 93-105. 10.1023/a:1015726608146
- Rogers NH, Perfield JW, 2nd, Strissel KJ, Obin MS, Greenberg AS (2009). Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology 150(5): 2161-2168. 10.1210/en.2008-1405
- Yakar S, Nunez NP, Pennisi P, Brodt P, Sun H, Fallavollita L, Zhao H, Scavo L, Novosyadlyy R, Kurshan N, Stannard B, East-Palmer J, Smith NC, Perkins SN, Fuchs-Young R, Barrett JC, Hursting SD, LeRoith D (2006). Increased tumor growth in mice with diet-induced obesity: impact of ovarian hormones. Endocrinology 147(12): 5826-5834. 10.1210/en.2006-0311
- Della Vedova MC, Munoz MD, Santillan LD, Plateo-Pignatari MG, Germano MJ, Rinaldi Tosi ME, Garcia S, Gomez NN, Fornes MW, Gomez Mejiba SE, Ramirez DC (2016). A Mouse Model of Diet-Induced Obesity Resembling Most Features of Human Metabolic Syndrome. Nutr Metab Insights 9: 93-102. 10.4137/NMI.S32907
- Tiwari P, Blank A, Cui C, Schoenfelt KQ, Zhou G, Xu Y, Khramtsova G, Olopade F, Shah AM, Khan SA, Rosner MR, Becker L (2019). Metabolically activated adipose tissue macrophages link obesity to triple-negative breast cancer. J Exp Med 216(6): 1345-1358. 10.1084/jem.20181616
- Molnar J, Yu S, Mzhavia N, Pau C, Chereshnev I, Dansky HM (2005). Diabetes induces endothelial dysfunction but does not increase neointimal formation in high-fat diet fed C57BL/6J mice. Circ Res 96(11): 1178-1184. 10.1161/01.RES.0000168634.74330.ed
- Halberg N, Wernstedt-Asterholm I, Scherer PE (2008). The adipocyte as an endocrine cell. Endocrinol Metab Clin North Am 37(3): 753-768. 10.1016/j.ecl.2008.07.002
- Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, et al. (1995). Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1(11): 1155-1161. 10.1038/nm1195-1155
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, et al. (1996). Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 334(5): 292-295. 10.1056/NEJM199602013340503
- Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS (1995). Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med 1(12): 1311-1314. 10.1038/nm1295-1311
- Myers MG, Jr., Leibel RL, Seeley RJ, Schwartz MW (2010). Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab 21(11): 643-651. 10.1016/j.tem.2010.08.002
- Sanchez-Jimenez F, Perez-Perez A, de la Cruz-Merino L, Sanchez-Margalet V (2019). Obesity and Breast Cancer: Role of Leptin. Front Oncol 9: 596. 10.3389/fonc.2019.00596
- Dutta D, Ghosh S, Pandit K, Mukhopadhyay P, Chowdhury S (2012). Leptin and cancer: Pathogenesis and modulation. Indian J Endocrinol Metab 16(Suppl 3): S596-600. 10.4103/2230-8210.105577
- Garofalo C, Koda M, Cascio S, Sulkowska M, Kanczuga-Koda L, Golaszewska J, Russo A, Sulkowski S, Surmacz E (2006). Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: possible role of obesity-related stimuli. Clin Cancer Res 12(5): 1447-1453. 10.1158/1078-0432.CCR-05-1913
- Ishikawa M, Kitayama J, Nagawa H (2004). Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin Cancer Res 10(13): 4325-4331. 10.1158/1078-0432.CCR-03-0749
- Jarde T, Perrier S, Vasson MP, Caldefie-Chezet F (2011). Molecular mechanisms of leptin and adiponectin in breast cancer. Eur J Cancer 47(1): 33-43. 10.1016/j.ejca.2010.09.005
- Endo H, Hosono K, Uchiyama T, Sakai E, Sugiyama M, Takahashi H, Nakajima N, Wada K, Takeda K, Nakagama H, Nakajima A (2011). Leptin acts as a growth factor for colorectal tumours at stages subsequent to tumour initiation in murine colon carcinogenesis. Gut 60(10): 1363-1371. 10.1136/gut.2010.235754
- Blanquer-Rossello MDM, Oliver J, Sastre-Serra J, Valle A, Roca P (2016). Leptin regulates energy metabolism in MCF-7 breast cancer cells. Int J Biochem Cell Biol 72: 18-26. 10.1016/j.biocel.2016.01.002
- Mahbouli S, Der Vartanian A, Ortega S, Rouge S, Vasson MP, Rossary A (2017). Leptin induces ROS via NOX5 in healthy and neoplastic mammary epithelial cells. Oncol Rep 38(5): 3254-3264. 10.3892/or.2017.6009
- Raut PK, Kim SH, Choi DY, Jeong GS, Park PH (2019). Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: Critical roles of estrogen receptor signaling and reactive oxygen species production. Biochem Pharmacol 161: 73-88. 10.1016/j.bcp.2019.01.006
- Wu MH, Chou YC, Chou WY, Hsu GC, Chu CH, Yu CP, Yu JC, Sun CA (2009). Circulating levels of leptin, adiposity and breast cancer risk. Br J Cancer 100(4): 578-582. 10.1038/sj.bjc.6604913
- Khabaz MN, Abdelrahman A, Butt N, Damnhory L, Elshal M, Aldahlawi AM, Ashoor S, Al-Maghrabi B, Dobson P, Brown B, Al-Sakkaf K, Al-Qahtani M, Al-Maghrabi J (2017). Immunohistochemical staining of leptin is associated with grade, stage, lymph node involvement, recurrence, and hormone receptor phenotypes in breast cancer. BMC Womens Health 17(1): 105. 10.1186/s12905-017-0459-y
- Zheng Q, Dunlap SM, Zhu J, Downs-Kelly E, Rich J, Hursting SD, Berger NA, Reizes O (2011). Leptin deficiency suppresses MMTV-Wnt-1 mammary tumor growth in obese mice and abrogates tumor initiating cell survival. Endocr Relat Cancer 18(4): 491-503. 10.1530/ERC-11-0102
- Bowers LW, Rossi EL, McDonell SB, Doerstling SS, Khatib SA, Lineberger CG, Albright JE, Tang X, deGraffenried LA, Hursting SD (2018). Leptin Signaling Mediates Obesity-Associated CSC Enrichment and EMT in Preclinical TNBC Models. Mol Cancer Res 16(5): 869-879. 10.1158/1541-7786.MCR-17-0508
- Chang CC, Wu MJ, Yang JY, Camarillo IG, Chang CJ (2015). Leptin-STAT3-G9a Signaling Promotes Obesity-Mediated Breast Cancer Progression. Cancer Res 75(11): 2375-2386. 10.1158/0008-5472.CAN-14-3076
- Murphy KA, James BR, Sjaastad FV, Kucaba TA, Kim H, Brincks EL, Chua SC, Jr., Wilber A, Griffith TS (2018). Cutting Edge: Elevated Leptin during Diet-Induced Obesity Reduces the Efficacy of Tumor Immunotherapy. J Immunol 201(7): 1837-1841. 10.4049/jimmunol.1701738
- Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, Ng MR, Nia HT, Grahovac J, Kao S, Babykutty S, Huang Y, Jung K, Rahbari NN, Han X, Chauhan VP, Martin JD, Kahn J, Huang P, Desphande V, Michaelson J, Michelakos TP, Ferrone CR, Soares R, Boucher Y, Fukumura D, Jain RK (2016). Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov 6(8): 852-869. 10.1158/2159-8290.CD-15-1177
- Halter SA, Dempsey P, Matsui Y, Stokes MK, Graves-Deal R, Hogan BL, Coffey RJ (1992). Distinctive patterns of hyperplasia in transgenic mice with mouse mammary tumor virus transforming growth factor-alpha. Characterization of mammary gland and skin proliferations. Am J Pathol 140(5): 1131-1146. 1316084
- Cleary MP, Phillips FC, Getzin SC, Jacobson TL, Jacobson MK, Christensen TA, Juneja SC, Grande JP, Maihle NJ (2003). Genetically obese MMTV-TGF-alpha/Lep(ob)Lep(ob) female mice do not develop mammary tumors. Breast Cancer Res Treat 77(3): 205-215. 10.1023/a:1021891825399
- Cleary MP, Juneja SC, Phillips FC, Hu X, Grande JP, Maihle NJ (2004). Leptin receptor-deficient MMTV-TGF-alpha/Lepr(db)Lepr(db) female mice do not develop oncogene-induced mammary tumors. Exp Biol Med 229(2): 182-193. 10.1177/153537020422900207
- Dalamaga M, Diakopoulos KN, Mantzoros CS (2012). The role of adiponectin in cancer: a review of current evidence. Endocr Rev 33(4): 547-594. 10.1210/er.2011-1015
- Berg AH, Combs TP, Du X, Brownlee M, Scherer PE (2001). The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 7(8): 947-953. 10.1038/90992
- Tumminia A, Vinciguerra F, Parisi M, Graziano M, Sciacca L, Baratta R, Frittitta L (2019). Adipose Tissue, Obesity and Adiponectin: Role in Endocrine Cancer Risk. Int J Mol Sci 20(12). 10.3390/ijms20122863
- Katira A, Tan PH (2016). Evolving role of adiponectin in cancer-controversies and update. Cancer Biol Med 13(1): 101-119. 10.28092/j.issn.2095-3941.2015.0092
- Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF (2004). T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A 101(28): 10308-10313. 10.1073/pnas.0403382101
- Parida S, Siddharth S, Sharma D (2019). Adiponectin, Obesity, and Cancer: Clash of the Bigwigs in Health and Disease. Int J Mol Sci 20(10)_ 2519. 10.3390/ijms20102519
- Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, Wade MR, Tenorio VM, Kuo MS, Brozinick JT, Zhang BB, Birnbaum MJ, Summers SA, Scherer PE (2011). Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17(1): 55-63. 10.1038/nm.2277
- Nagahashi M, Yamada A, Katsuta E, Aoyagi T, Huang WC, Terracina KP, Hait NC, Allegood JC, Tsuchida J, Yuza K, Nakajima M, Abe M, Sakimura K, Milstien S, Wakai T, Spiegel S, Takabe K (2018). Targeting the SphK1/S1P/S1PR1 Axis That Links Obesity, Chronic Inflammation, and Breast Cancer Metastasis. Cancer Res 78(7): 1713-1725. 10.1158/0008-5472.CAN-17-1423
- Wei EK, Giovannucci E, Fuchs CS, Willett WC, Mantzoros CS (2005). Low plasma adiponectin levels and risk of colorectal cancer in men: a prospective study. J Natl Cancer Inst 97(22): 1688-1694. 10.1093/jnci/dji376
- Mutoh M, Teraoka N, Takasu S, Takahashi M, Onuma K, Yamamoto M, Kubota N, Iseki T, Kadowaki T, Sugimura T, Wakabayashi K (2011). Loss of adiponectin promotes intestinal carcinogenesis in Min and wild-type mice. Gastroenterology 140(7): 2000-2008. 10.1053/j.gastro.2011.02.019
- Fujisawa T, Endo H, Tomimoto A, Sugiyama M, Takahashi H, Saito S, Inamori M, Nakajima N, Watanabe M, Kubota N, Yamauchi T, Kadowaki T, Wada K, Nakagama H, Nakajima A (2008). Adiponectin suppresses colorectal carcinogenesis under the high-fat diet condition. Gut 57(11): 1531-1538. 10.1136/gut.2008.159293
- Saxena NK, Fu PP, Nagalingam A, Wang J, Handy J, Cohen C, Tighiouart M, Sharma D, Anania FA (2010). Adiponectin modulates C-jun N-terminal kinase and mammalian target of rapamycin and inhibits hepatocellular carcinoma. Gastroenterology 139(5): 1762-1773. 10.1053/j.gastro.2010.07.001
- Man K, Ng KT, Xu A, Cheng Q, Lo CM, Xiao JW, Sun BS, Lim ZX, Cheung JS, Wu EX, Sun CK, Poon RT, Fan ST (2010). Suppression of liver tumor growth and metastasis by adiponectin in nude mice through inhibition of tumor angiogenesis and downregulation of Rho kinase/IFN-inducible protein 10/matrix metalloproteinase 9 signaling. Clin Cancer Res 16(3): 967-977. 10.1158/1078-0432.CCR-09-1487
- Brakenhielm E, Veitonmaki N, Cao R, Kihara S, Matsuzawa Y, Zhivotovsky B, Funahashi T, Cao Y (2004). Adiponectin-induced antiangiogenesis and antitumor activity involve caspase-mediated endothelial cell apoptosis. Proc Natl Acad Sci U S A 101(8): 2476-2481. 10.1073/pnas.0308671100
- Landskroner-Eiger S, Qian B, Muise ES, Nawrocki AR, Berger JP, Fine EJ, Koba W, Deng Y, Pollard JW, Scherer PE (2009). Proangiogenic contribution of adiponectin toward mammary tumor growth in vivo. Clin Cancer Res 15(10): 3265-3276. 10.1158/1078-0432.CCR-08-2649
- Denzel MS, Hebbard LW, Shostak G, Shapiro L, Cardiff RD, Ranscht B (2009). Adiponectin deficiency limits tumor vascularization in the MMTV-PyV-mT mouse model of mammary cancer. Clin Cancer Res 15(10): 3256-3264. 10.1158/1078-0432.CCR-08-2661
- Gui Y, Pan Q, Chen X, Xu S, Luo X, Chen L (2017). The association between obesity related adipokines and risk of breast cancer: a meta-analysis. Oncotarget 8(43): 75389-75399. 10.18632/oncotarget.17853
- Jamaluddin MS, Weakley SM, Yao Q, Chen C (2012). Resistin: functional roles and therapeutic considerations for cardiovascular disease. Br J Pharmacol 165(3): 622-632. 10.1111/j.1476-5381.2011.01369.x
- Gao Y, Chen X, He Q, Gimple RC, Liao Y, Wang L, Wu R, Xie Q, Rich JN, Shen K, Yuan Z (2020). Adipocytes promote breast tumorigenesis through TAZ-dependent secretion of Resistin. Proc Natl Acad Sci U S A 117(52): 33295-33304. 10.1073/pnas.2005950117
- Qureshi R, Picon-Ruiz M, Aurrekoetxea-Rodriguez I, Nunes de Paiva V, D’Amico M, Yoon H, Radhakrishnan R, Morata-Tarifa C, Ince T, Lippman ME, Thaller SR, Rodgers SE, Kesmodel S, Vivanco MDM, Slingerland JM (2020). The Major Pre- and Postmenopausal Estrogens Play Opposing Roles in Obesity-Driven Mammary Inflammation and Breast Cancer Development. Cell Metab 31(6): 1154-1172 e1159. 10.1016/j.cmet.2020.05.008
- Nunez NP, Oh WJ, Rozenberg J, Perella C, Anver M, Barrett JC, Perkins SN, Berrigan D, Moitra J, Varticovski L, Hursting SD, Vinson C (2006). Accelerated tumor formation in a fatless mouse with type 2 diabetes and inflammation. Cancer Res 66(10): 5469-5476. 10.1158/0008-5472.CAN-05-4102
- Pollak M (2012). The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 12(3): 159-169. 10.1038/nrc3215
- Pollak MN, Schernhammer ES, Hankinson SE (2004). Insulin-like growth factors and neoplasia. Nat Rev Cancer 4(7): 505-518. 10.1038/nrc1387
- Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M (2010). Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer 17(2): 351-360. 10.1677/ERC-09-0252
- Cox ME, Gleave ME, Zakikhani M, Bell RH, Piura E, Vickers E, Cunningham M, Larsson O, Fazli L, Pollak M (2009). Insulin receptor expression by human prostate cancers. Prostate 69(1): 33-40. 10.1002/pros.20852
- Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, Park E, Gee JM, Finlay P, Jones HE, Nicholson RI, Carboni J, Gottardis M, Pollak M, Dunn SE (2008). Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res 68(24): 10238-10246. 10.1158/0008-5472.CAN-08-2755
- Kalli KR, Falowo OI, Bale LK, Zschunke MA, Roche PC, Conover CA (2002). Functional insulin receptors on human epithelial ovarian carcinoma cells: implications for IGF-II mitogenic signaling. Endocrinology 143(9): 3259-3267. 10.1210/en.2001-211408
- Wang CF, Zhang G, Zhao LJ, Qi WJ, Li XP, Wang JL, Wei LH (2013). Overexpression of the insulin receptor isoform A promotes endometrial carcinoma cell growth. PLoS One 8(8): e69001. 10.1371/journal.pone.0069001
- Perry RJ, Shulman GI (2020). Mechanistic Links between Obesity, Insulin, and Cancer. Trends Cancer 6(2): 75-78. 10.1016/j.trecan.2019.12.003
- Gallagher EJ, LeRoith D (2020). Hyperinsulinaemia in cancer. Nat Rev Cancer 20(11): 629-644. 10.1038/s41568-020-0295-5
- Rabin-Court A, Rodrigues MR, Zhang XM, Perry RJ (2019). Obesity-associated, but not obesity-independent, tumors respond to insulin by increasing mitochondrial glucose oxidation. PLoS One 14(6): e0218126. 10.1371/journal.pone.0218126
- Nasiri AR, Rodrigues MR, Li Z, Leitner BP, Perry RJ (2019). SGLT2 inhibition slows tumor growth in mice by reversing hyperinsulinemia. Cancer Metab 7: 10. 10.1186/s40170-019-0203-1
- Bhowmick NA, Neilson EG, Moses HL (2004). Stromal fibroblasts in cancer initiation and progression. Nature 432(7015): 332-337. 10.1038/nature03096
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE (2009). Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 29(16): 4467-4483. 10.1128/MCB.00192-09
- Druso JE, Fischbach C (2018). Biophysical Properties of Extracellular Matrix: Linking Obesity and Cancer. Trends Cancer 4(4): 271-273. 10.1016/j.trecan.2018.02.001
- Seo BR, Bhardwaj P, Choi S, Gonzalez J, Andresen Eguiluz RC, Wang K, Mohanan S, Morris PG, Du B, Zhou XK, Vahdat LT, Verma A, Elemento O, Hudis CA, Williams RM, Gourdon D, Dannenberg AJ, Fischbach C (2015). Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci Transl Med 7(301): 301ra130. 10.1126/scitranslmed.3010467
- Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, Rasmiena AA, Kaur S, Gulati T, Goh PK, Treloar AE, Archer S, Brown WA, Muller M, Watt MJ, Ohara O, McLean CA, Tiganis T (2018). Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 175(5): 1289-1306 e1220. 10.1016/j.cell.2018.09.053
- Wishart AL, Conner SJ, Guarin JR, Fatherree JP, Peng Y, McGinn RA, Crews R, Naber SP, Hunter M, Greenberg AS, Oudin MJ (2020). Decellularized extracellular matrix scaffolds identify full-length collagen VI as a driver of breast cancer cell invasion in obesity and metastasis. Sci Adv 6(43): eabc3175. 10.1126/sciadv.abc3175
- Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, Lee H, Temple K, Graves R, Pollard J, Chopra N, Russell RG, Sasisekharan R, Trock BJ, Lippman M, Calvert VS, Petricoin EF, 3rd, Liotta L, Dadachova E, Pestell RG, Lisanti MP, Bonaldo P, Scherer PE (2005). Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Invest 115(5): 1163-1176. 10.1172/JCI23424
- Springer NL, Iyengar NM, Bareja R, Verma A, Jochelson MS, Giri DD, Zhou XK, Elemento O, Dannenberg AJ, Fischbach C (2019). Obesity-Associated Extracellular Matrix Remodeling Promotes a Macrophage Phenotype Similar to Tumor-Associated Macrophages. Am J Pathol 189(10): 2019-2035. 10.1016/j.ajpath.2019.06.005
- Hermano E, Goldberg R, Rubinstein AM, Sonnenblick A, Maly B, Nahmias D, Li JP, Bakker MAH, van der Vlag J, Vlodavsky I, Peretz T, Elkin M (2019). Heparanase Accelerates Obesity-Associated Breast Cancer Progression. Cancer Res 79(20): 5342-5354. 10.1158/0008-5472.CAN-18-4058
- Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, Nieman KM, Pascual G, Benitah SA, Montag A, Yamada SD, Abumrad NA, Lengyel E (2018). Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 37(17): 2285-2301. 10.1038/s41388-017-0093-z
- Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, Bescos C, Di Croce L, Benitah SA (2017). Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541(7635): 41-45. 10.1038/nature20791
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E (2011). Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17(11): 1498-1503. 10.1038/nm.2492
- Hao J, Zhang Y, Yan X, Yan F, Sun Y, Zeng J, Waigel S, Yin Y, Fraig MM, Egilmez NK, Suttles J, Kong M, Liu S, Cleary MP, Sauter E, Li B (2018). Circulating Adipose Fatty Acid Binding Protein Is a New Link Underlying Obesity-Associated Breast/Mammary Tumor Development. Cell Metab 28(5): 689-705 e685. 10.1016/j.cmet.2018.07.006
- Madak-Erdogan Z, Band S, Zhao YC, Smith BP, Kulkoyluoglu-Cotul E, Zuo Q, Santaliz Casiano A, Wrobel K, Rossi G, Smith RL, Kim SH, Katzenellenbogen JA, Johnson ML, Patel M, Marino N, Storniolo AMV, Flaws JA (2019). Free Fatty Acids Rewire Cancer Metabolism in Obesity-Associated Breast Cancer via Estrogen Receptor and mTOR Signaling. Cancer Res 79(10): 2494-2510. 10.1158/0008-5472.CAN-18-2849
- Ringel AE, Drijvers JM, Baker GJ, Catozzi A, Garcia-Canaveras JC, Gassaway BM, Miller BC, Juneja VR, Nguyen TH, Joshi S, Yao CH, Yoon H, Sage PT, LaFleur MW, Trombley JD, Jacobson CA, Maliga Z, Gygi SP, Sorger PK, Rabinowitz JD, Sharpe AH, Haigis MC (2020). Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 183(7): 1848-1866 e1826. 10.1016/j.cell.2020.11.009
- Wang D, Bi Y, Hu L, Luo Y, Ji J, Mao AZ, Logsdon CD, Li E, Abbruzzese JL, Li Z, Yang VW, Lu W (2019). Obesogenic high-fat diet heightens aerobic glycolysis through hyperactivation of oncogenic KRAS. Cell Commun Signal 17(1): 19. 10.1186/s12964-019-0333-7
- Zaytouni T, Tsai PY, Hitchcock DS, DuBois CD, Freinkman E, Lin L, Morales-Oyarvide V, Lenehan PJ, Wolpin BM, Mino-Kenudson M, Torres EM, Stylopoulos N, Clish CB, Kalaany NY (2017). Critical role for arginase 2 in obesity-associated pancreatic cancer. Nat Commun 8(1): 242. 10.1038/s41467-017-00331-y
- Ramos CA, Ouyang C, Qi Y, Chung Y, Cheng CT, LaBarge MA, Seewaldt VL, Ann DK (2020). A Non-canonical Function of BMAL1 Metabolically Limits Obesity-Promoted Triple-Negative Breast Cancer. iScience 23(2): 100839. 10.1016/j.isci.2020.100839
- Apostolopoulos V, de Courten MP, Stojanovska L, Blatch GL, Tangalakis K, de Courten B (2016). The complex immunological and inflammatory network of adipose tissue in obesity. Mol Nutr Food Res 60(1): 43-57. 10.1002/mnfr.201500272
- Incio J, Tam J, Rahbari NN, Suboj P, McManus DT, Chin SM, Vardam TD, Batista A, Babykutty S, Jung K, Khachatryan A, Hato T, Ligibel JA, Krop IE, Puchner SB, Schlett CL, Hoffmman U, Ancukiewicz M, Shibuya M, Carmeliet P, Soares R, Duda DG, Jain RK, Fukumura D (2016). PlGF/VEGFR-1 Signaling Promotes Macrophage Polarization and Accelerated Tumor Progression in Obesity. Clin Cancer Res 22(12): 2993-3004. 10.1158/1078-0432.CCR-15-1839
- Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D, Gomez SB, Ji B, Huang H, Wang H, Fleming JB, Logsdon CD, Cruz-Monserrate Z (2013). A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 145(6): 1449-1458. 10.1053/j.gastro.2013.08.018
- Khasawneh J, Schulz MD, Walch A, Rozman J, Hrabe de Angelis M, Klingenspor M, Buck A, Schwaiger M, Saur D, Schmid RM, Kloppel G, Sipos B, Greten FR, Arkan MC (2009). Inflammation and mitochondrial fatty acid beta-oxidation link obesity to early tumor promotion. Proc Natl Acad Sci U S A 106(9): 3354-3359. 10.1073/pnas.0802864106
- Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M (2010). Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140(2): 197-208. 10.1016/j.cell.2009.12.052
- Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, Koike K, Kaufman RJ, Karin M (2014). ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26(3): 331-343. 10.1016/j.ccr.2014.07.001
- Schulz MD, Atay C, Heringer J, Romrig FK, Schwitalla S, Aydin B, Ziegler PK, Varga J, Reindl W, Pommerenke C, Salinas-Riester G, Bock A, Alpert C, Blaut M, Polson SC, Brandl L, Kirchner T, Greten FR, Polson SW, Arkan MC (2014). High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 514(7523): 508-512. 10.1038/nature13398
- Bousquenaud M, Fico F, Solinas G, Ruegg C, Santamaria-Martinez A (2018). Obesity promotes the expansion of metastasis-initiating cells in breast cancer. Breast Cancer Res 20(1): 104. 10.1186/s13058-018-1029-4
- James BR, Tomanek-Chalkley A, Askeland EJ, Kucaba T, Griffith TS, Norian LA (2012). Diet-induced obesity alters dendritic cell function in the presence and absence of tumor growth. J Immunol 189(3): 1311-1321. 10.4049/jimmunol.1100587
- Wogsland CE, Lien HE, Pedersen L, Hanjra P, Grondal SM, Brekken RA, Lorens JB, Halberg N (2020). High dimensional immunotyping of the obese tumor microenvironment reveals model specific adaptation. bioRxiv. 10.1101/2020.10.14.338806
- Wunderlich CM, Ackermann PJ, Ostermann AL, Adams-Quack P, Vogt MC, Tran ML, Nikolajev A, Waisman A, Garbers C, Theurich S, Mauer J, Hovelmeyer N, Wunderlich FT (2018). Obesity exacerbates colitis-associated cancer via IL-6-regulated macrophage polarisation and CCL-20/CCR-6-mediated lymphocyte recruitment. Nat Commun 9(1): 1646. 10.1038/s41467-018-03773-0
- Hayashi T, Fujita K, Nojima S, Hayashi Y, Nakano K, Ishizuya Y, Wang C, Yamamoto Y, Kinouchi T, Matsuzaki K, Jingushi K, Kato T, Kawashima A, Nagahara A, Ujike T, Uemura M, Pena M, Gordetsky JB, Morii E, Tsujikawa K, Netto GJ, Nonomura N (2018). High-Fat Diet-Induced Inflammation Accelerates Prostate Cancer Growth via IL6 Signaling. Clin Cancer Res 24(17): 4309-4318. 10.1158/1078-0432.CCR-18-0106
- Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM, Xie Q, Markan KR, Li W, Potthoff MJ, Fuentes-Mattei E, Ellies LG, Knudson CM, Lee MH, Yeung SJ, Cassel SL, Sutterwala FS, Zhang W (2016). Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun 7: 13007. 10.1038/ncomms13007
- Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, Mirsoian A, Minnar CM, Stoffel KM, Sturgill IR, Grossenbacher SK, Withers SS, Rebhun RB, Hartigan-O’Connor DJ, Mendez-Lagares G, Tarantal AF, Isseroff RR, Griffith TS, Schalper KA, Merleev A, Saha A, Maverakis E, Kelly K, Aljumaily R, Ibrahimi S, Mukherjee S, Machiorlatti M, Vesely SK, Longo DL, Blazar BR, et al. (2019). Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med 25(1): 141-151. 10.1038/s41591-018-0221-5
- McQuade JL, Daniel CR, Hess KR, Mak C, Wang DY, Rai RR, Park JJ, Haydu LE, Spencer C, Wongchenko M, Lane S, Lee DY, Kaper M, McKean M, Beckermann KE, Rubinstein SM, Rooney I, Musib L, Budha N, Hsu J, Nowicki TS, Avila A, Haas T, Puligandla M, Lee S, Fang S, Wargo JA, Gershenwald JE, Lee JE, Hwu P, et al. (2018). Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol 19(3): 310-322. 10.1016/S1470-2045(18)30078-0
- Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y, Nakajima-Takagi Y, Iwama A, Koga T, Sugimoto Y, Ozawa T, Nakamura M, Kumagai M, Watashi K, Taketo MM, Aoki T, Narumiya S, Oshima M, Arita M, Hara E, Ohtani N (2017). Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov 7(5): 522-538. 10.1158/2159-8290.CD-16-0932
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444(7122): 1022-1023. 10.1038/4441022a
- Turnbaugh PJ, Backhed F, Fulton L, Gordon JI (2008). Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3(4): 213-223. 10.1016/j.chom.2008.02.015
- Morrison DJ, Preston T (2016). Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 7(3): 189-200. 10.1080/19490976.2015.1134082
- Cani PD, Jordan BF (2018). Gut microbiota-mediated inflammation in obesity: a link with gastrointestinal cancer. Nat Rev Gastroenterol Hepatol 15(11): 671-682. 10.1038/s41575-018-0025-6
- Ling C, Ronn T (2019). Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab 29(5): 1028-1044. 10.1016/j.cmet.2019.03.009
- Labbe DP, Zadra G, Yang M, Reyes JM, Lin CY, Cacciatore S, Ebot EM, Creech AL, Giunchi F, Fiorentino M, Elfandy H, Syamala S, Karoly ED, Alshalalfa M, Erho N, Ross A, Schaeffer EM, Gibb EA, Takhar M, Den RB, Lehrer J, Karnes RJ, Freedland SJ, Davicioni E, Spratt DE, Ellis L, Jaffe JD, D’Amico AV, Kantoff PW, Bradner JE, et al. (2019). High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program. Nat Commun 10(1): 4358. 10.1038/s41467-019-12298-z
- Rossi EL, Dunlap SM, Bowers LW, Khatib SA, Doerstling SS, Smith LA, Ford NA, Holley D, Brown PH, Estecio MR, Kusewitt DF, deGraffenried LA, Bultman SJ, Hursting SD (2017). Energy Balance Modulation Impacts Epigenetic Reprogramming, ERalpha and ERbeta Expression, and Mammary Tumor Development in MMTV-neu Transgenic Mice. Cancer Res 77(9): 2500-2511. 10.1158/0008-5472.CAN-16-2795
- Reid MA, Dai Z, Locasale JW (2017). The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol 19(11): 1298-1306. 10.1038/ncb3629
- Nogueira LM, Dunlap SM, Ford NA, Hursting SD (2012). Calorie restriction and rapamycin inhibit MMTV-Wnt-1 mammary tumor growth in a mouse model of postmenopausal obesity. Endocr Relat Cancer 19(1):57-68. 10.1530/ERC-11-0213
- Zhang Y, Daquinag AC, Amaya-Manzanares F, Sirin O, Tseng C, Kolonin MG (2012). Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res 72(20):5198-208. 10.1158/0008-5472.CAN-12-0294
- O’Flanagan CH, Rossi EL, McDonell SB, Chen X, Tsai YH, Parker JS, Usary J, Perou CM, Hursting SD (2017). Metabolic reprogramming underlies metastatic potential in an obesity-responsive murine model of metastatic triple negative breast cancer. NPJ Breast Cancer 3:26. 10.1038/s41523-017-0027-5
- Day SD, Enos RT, McClellan JL, Steiner JL, Velázquez KT, Murphy EA (2013). Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine 64(1):454-62. 10.1016/j.cyto.2013.04.031
- Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y, Nakajima-Takagi Y, Iwama A, Koga T, Sugimoto Y, Ozawa T, Nakamura M, Kumagai M, Watashi K, Taketo MM, Aoki T, Narumiya S, Oshima S, Arita M, Hara E, Ohtani N (2017). Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov 7(5):522-538. 10.1158/2159-8290.CD-16-0932
- Li Z, Liu H, He J, Wang Z, Yin Z, You G, WangZ1, Davis RE, Lin P, Bergsagel PL, Manasanch EE, Wong STC, Esnaola NF, Chang JC, Orlowski RZ, Yi Q, Yang J (2021). Acetyl-CoA Synthetase 2: A Critical Linkage in Obesity-Induced Tumorigenesis in Myeloma. Cell Metab 33(1):78-93.e7. 10.1016/j.cmet.2020.12.011
- Zyromski NJ, Mathur A, Pitt HA, Wade TE, Wang S, Nakshatri P, Swartz-Basile DA, Nakshatri H (2009). Obesity potentiates the growth and dissemination of pancreatic cancer. Surgery 146(2):258-63. 10.1016/j.surg.2009.02.024
- Dawson DW, Hertzer K, Moro A, Donald G, Chang HH, Go VL, Pandol SJ, Lugea A, Gukovskaya AS, Li G, Hines OJ, Rozengurt E, Eibl G (2013). High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev Res 6(10):1064-73. 10.1158/1940-6207.CAPR-13-0065
- Gomez-Chou SB, Swidnicka-Siergiejko AK, Badi N, Chavez-Tomar M, Lesinski GB, Bekaii-Saab T, Farren MR, Mace TA, Schmidt C, Liu Y, Deng D, Hwang RF, Zhou L, Moore T, Chatterjee D, Wang H, Leng X, Arlinghaus RB, Logsdon CD, Cruz-Monserrate Z (2017). Lipocalin-2 Promotes Pancreatic Ductal Adenocarcinoma by Regulating Inflammation in the Tumor Microenvironment. Cancer Res 77(10):2647-2660. 10.1158/0008-5472.CAN-16-1986
- Chang HH, Moro A, Takakura K, Su HY, Mo A, Nakanishi M, Waldron RT, French SW, Dawson DW, Hines OJ, Li G, Go VLW, Sinnett-Smith J, Pandol SJ, Lugea A, Gukovskaya AS, Duff MO, Rosenberg DW, Rozengurt E, Eibl G (2017). Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS One 12(9):e0184455. 10.1371/journal.pone.0184455
- Sasaki A, Nagatake T, Egami R, Gu G, Takigawa I, Ikeda W, Nakatani T, Kunisawa J, Fujita Y (2018). Obesity Suppresses Cell-Competition-Mediated Apical Elimination of RasV12-Transformed Cells from Epithelial Tissues. Cell Rep 23(4):974-982. 10.1016/j.celrep.2018.03.104
- Luo Y, Yang Y, Liu M, Wang D, Wang F, Bi Y, Ji J, Li S, Liu Y, Chen R, Huang H, Wang X, Swidnicka-Siergiejko AK, Janowitz T, Beyaz S, Wang G, Xu S, Bialkowska AB, Luo CK, Pin CL, Liang G, Lu X, Wu M, Shroyer KR, Wolff RA, Plunkett W, Ji B, Li Z, Li E, Li X, Yang VW, Logsdon CD, Abbruzzese JL, Lu W (2019). Oncogenic KRAS Reduces Expression of FGF21 in Acinar Cells to Promote Pancreatic Tumorigenesis in Mice on a High-Fat Diet. Gastroenterology 157(5):1413-1428.e11. 10.1053/j.gastro.2019.07.030
- Kwon OJ, Zhang B, Zhang L, Xin L (2016). High fat diet promotes prostatic basal-to-luminal differentiation and accelerates initiation of prostate epithelial hyperplasia originated from basal cells. Stem Cell Res 16(3):682-91. 10.1016/j.scr.2016.04.009
–
ACKNOWLEDGMENTS
We thank members of our laboratory and Anders Molven for insightful discussions. NH was supported by a Trond Mohn Foundation Starter grant, The Norwegian Research Council (#275250) and the Norwegian Cancer Society (#207028). We apologize to colleagues whose important work was not cited due to space limitations. Figure 1 was created using Biorender.
COPYRIGHT
© 2021

Cellular mechanisms linking cancers to obesity by Liu et al. is licensed under a Creative Commons Attribution 4.0 International License.


