Back to article: Mitochondria, mitophagy, and metabolic disease: towards assembling the puzzle
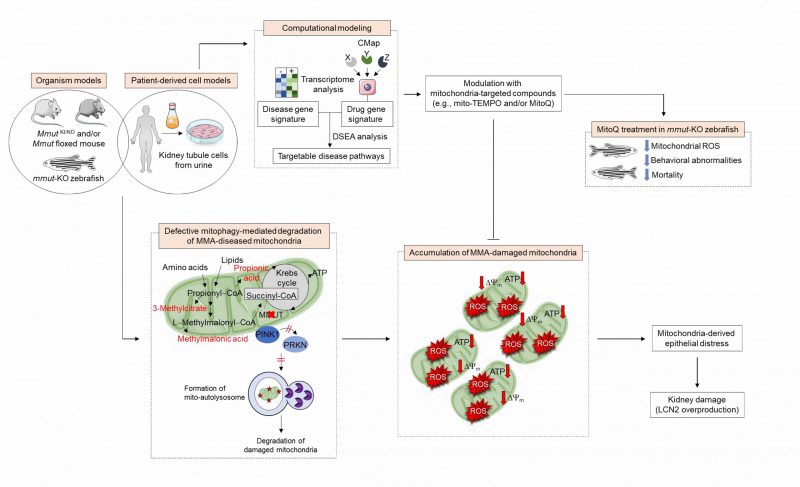
FIGURE 1: Pathophysiology and identification of druggable targets in methylmalonic acidemia. In MMA-affected kidney cells and zebrafish, deficiency of the enzyme MMUT and the resulting accumulation of toxic organic acids trigger mitochondrial alterations that are characterized by a collapse of the mitochondrial membrane potential (Δψm), abnormal bioenergetics profiling, and increased generation of mitochondrial ROS and oxidative stress. Faulty execution of PINK1-PRKN-mediated mitophagy induced by MMUT deficiency impedes the delivery of damaged mitochondria and their dismantling by autophagy-lysosome degradation systems. This leads to the accumulation of dysfunctional, ROS-overproducing mitochondria that ultimately trigger epithelial distress (as evidenced by LCN2 overproduction) in patient-derived cells and disease-relevant phenotypes (as testified by liver/kidney mitochondriopathy, behavioral abnormalities and an excess of mortality) in mmut-deficient zebrafish. Unbiased drug–disease network perturbation modelling, based on transcriptome-wide profiles from MMA patient-derived kidney cells against a large compendium of gene signatures derived from Connectivity Map (CMAP) library of 1309 small bioactive drug compounds 1309 small bioactive drug compounds, identifies targetable disease-relevant biological pathways. Modulation of these identified targets (e.g., treatment with mitochondria-targeted ROS scavengers mito-TEMPO or MitoQ) repairs mitochondrial dysfunctions, neutralizes epithelial stress and cell damage in MMA cells, and improves disease–relevant phenotypes in mmut–deficient zebrafish.