Reviews:
Cell Stress, Vol. 3, No. 6, pp. 181 - 194; doi: 10.15698/cst2019.06.189
Remodeling of mitochondrial morphology and function: an emerging hallmark of cellular reprogramming

1 Department of Cell & Developmental Biology, Vanderbilt University, Nashville, TN 37240.
2 Neuroscience Program, Vanderbilt University Medical Center, Nashville, TN 37240.
3 Vanderbilt Brain Institute, Vanderbilt University Medical Center, Nashville, TN 37240.
4 Vanderbilt Center for Stem Cell Biology, Vanderbilt University, Nashville, TN 37240.
Keywords: BCL-2 family, mitochondria, mitochondrial dynamics, apoptosis.
Abbreviatons:
BAK – BCL-2 antagonist killer,
BAX – BCL-2 associated X,
BCL-2 – B-Cell Lymphoma-2,
BID – BCL-2 interacting domain death agonist,
DRP – dynamin-related GTPase protein,
ER – endoplasmic reticulum,
ESC – embryonic stem cell,
hPSC – human PSC,
iPSC – induced PSC,
OXPHOS – oxidative phosphorylation,
PSC - pluripotent stem cell,
ROCK - Rho kinase,
ROS – reactive oxygen species,
SAM – S-adenosylmethionine,
TCA – tricarboxylic acid.
Received originally: 30/12/2018 Received in revised form: 19/04/2019
Accepted: 30/05/2019
Published: 27/05/2019
Correspondence:
Vivian Gama, Ph.D., PMB407935; 465 21st Avenue South, 4150A, Vanderbilt University, Nashville, TN 37240-7935, Tel: (615) 875-9490; vivian.gama@vanderbilt.edu
Conflict of interest statement: Authors declare no conflict of interest.
Please cite this article as: Anuj Rastogi, Ela Contreras, Piyush Joshi and Vivian Gama (2019). Remodeling of mitochondrial morphology and function: an emerging hallmark of cellular reprogramming. Cell Stress 3(6): 181-194. doi: 10.15698/cst2019.06.189
Abstract
Research in the stem cell field has traditionally focused on understanding key transcriptional factors that provide pluripotent cell identity. However, much less is known about other critical non-transcriptional signaling networks that govern stem cell identity. Although we continue to gain critical insights into the mechanisms underlying mitochondrial morphology and function during cellular reprogramming – the process of reverting the fate of a differentiated cell into a stem cell, many uncertainties remain. Recent studies suggest an emerging landscape in which mitochondrial morphology and function have an active role in maintaining and regulating changes in cell identity. In this review, we will focus on these emerging concepts as crucial modulators of cellular reprogramming. Recognition of the widespread applicability of these concepts will increase our understanding of the mitochondrial mechanisms involved in cell identity, cell fate and disease.
INTRODUCTION
A paradigm shift from considering mitochondria mere powerhouses of the cell to realizing that they are a major regulatory hub began two decades ago with two discoveries: (1) that cytochrome c, a protein known previously only as an electron transporter, triggers cell disassembly during apoptosis once released into the cytoplasm [1]; and (2) that the B-Cell Lymphoma-2 (BCL-2) family controls this release [2]. Another shift followed from discovering that mitochondria are highly dynamic organelles, which fuse into each other and divide rapidly and reversibly in response to internal and external environmental cues. Subsequent discoveries established that mitochondrial fusion and fission (division), which are collectively known as mitochondrial dynamics [3, 4], are involved in aging [5], neurodegenerative diseases [6], and tumorigenesis [7][8][9]. Recent studies provide compelling evidence for a novel function of mitochondrial dynamics in human pluripotent stem cells (hPSCs). These investigations have revealed that the BCL-2 family is essential in hPSCs not only during cell death, but also through its ability to regulate the mitochondrial network, which is key for other cell fate decisions, such as self-renewal and pluripotency [10][11][12].
–
Stem cells can be divided into three main types: embryonic stem cells (ESCs), adult stem cells, and induced pluripotent stem cells (iPSCs). ESCs and iPSCs are defined by two key properties: self-renewal (defined as the ability to proliferate without lineage commitment) and pluripotency (defined as the ability to differentiate into the three main tissue lineages). Since the first human ESC (hESC) line was derived in 1998, hESCs have become efficient tools to study processes of human development and various aspects of human diseases, like cancer [13]. Since cells from the inner cell mass, from which ESCs are derived, can give rise to all tissues derived from germ layers (endoderm, mesoderm, and ectoderm), genomic instability is especially risky for their self-renewal and organ specific differentiation. Thus, not surprisingly, these cells are endowed with exquisite mechanisms to respond rapidly to apoptosis-inducing stress. Various studies have provided evidence of changes in apoptosis regulation, mitochondrial dynamics and metabolic function and regulation during the process of reprogramming (i.e. reverting a differentiated cell into a stem-like state, through the expression of master pluripotency transcription factors such as, OCT4 (Octamer-binding transcription factor 4), SOX2 (SRY(sex-determining region Y)-box 2), KLF4 (Kruppel-like factor 4) and c-MYC, collectively known as OSKM) [14][15].
–
Mitochondria-related hallmarks of reprogramming are beginning to emerge. In this review, we specifically discuss three biological capabilities acquired during the multistep transition of a somatic cell into a pluripotent stem cell. They include: inherent sensitivity to apoptosis, changes in mitochondrial morphology and localization, and a modified functional state of the mitochondria (i.e changes in metabolic requirements) [10][11][13][14][15][16][17] (Figure 1). In the following sections, we will explore the known underlying mechanisms involved in these changes highlighting recent discoveries as well as describe areas that are open to more detailed investigation. While we propose that these three mitochondria-related hallmarks constitute a requirement for reverting a differentiated cell into a stem cell, there are other mitochondrial events such as mitochondrial biogenesis, mitochondrial trafficking and mitochondrial transcription that also undergo dramatic changes during cellular reprogramming and that could also be essential for successfully reverting cell fate. Additional studies are needed to elucidate the impact of these events on the reprogramming process.
–
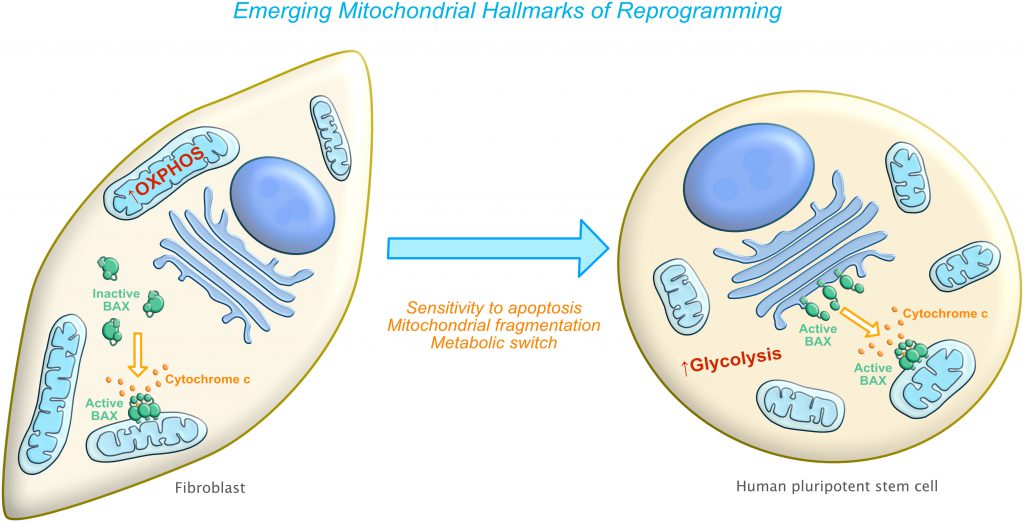 | FIGURE 1: Emerging mitochondria-related hallmarks of reprogramming. Left side: Differentiated cells (such as fibroblasts) have an elongated mitochondrial network with a preference for oxidative phosphorylation (OXPHOS) as a source of energy and are relatively resistant to apoptosis stimuli. Right side: After reprogramming, cells now have a fragmented mitochondrial network with a switch to using glycolysis as a preferred bioenergetic source. Cells become increasingly sensitive to cell death due to at least two main mechanisms: increased mitochondrial priming and pre-activated BAX. |
INCREASED SENSITIVITY TO CELL DEATH
The apoptotic process involves morphologic changes including cell constriction, chromatin condensation, nuclear envelope disruption followed by nucleus breakdown to discrete bodies, plasma membrane blebs formation, and finally the break-up of the cell into apoptotic bodies [18]. Activated cysteine proteases known as caspases cleave many vital cellular proteins (nuclear scaffold, cytoskeleton, etc.) followed by nuclear DNA degradation [19][20]. Caspase dependent apoptosis is classified into extrinsic and intrinsic pathways, mediated by death ligands and mitochondria respectively [21]. Our focus in this review will be on the mitochondrial pathway of apoptosis (Figure 2). The extrinsic pathway of apoptosis has been described extensively in several excellent reviews [22][23][24].
–
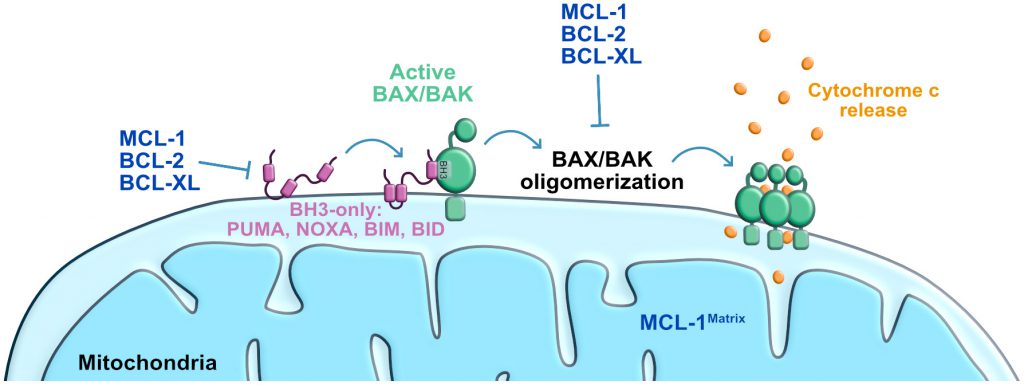 | FIGURE 2: Scheme of the mitochondrial pathway of apoptosis. Stress-induced activation of BAX/BAK leading to mitochondrial outer membrane permeabilization and cytochrome c release to the cytosol. Anti-apoptotic proteins block BAK/BAX activation/oligomerization induced by BH3-only proteins. BCL-XL: B-cell lymphoma extra-large; MCL-1: myeloid cell leukemia 1; BID: BH3-interacting domain death agonist; BIM: BCL-2 interacting mediator of cell death; PUMA: P53-upregulated modulator of apoptosis. |
–
The intrinsic apoptosis pathway, also known as the mitochondria mediated cell death pathway, is a form of regulated cell death initiated by a variety of perturbations including DNA damage [24]. These stresses induce the activation of the transcription factor P53 that then initiates a response to arrest cell cycle and repair DNA [25][26]. If repair is not achieved, an apoptosis response is triggered leading to the activation of the BCL-2 associated X (BAX) and/or BCL-2 antagonist killer 1 (BAK1; best known as BAK), these pro-apoptotic effector proteins of the BCL-2 family then permeabilize the outer mitochondrial membrane [27][28][29][30][31][32][33]. BAX and BAK can be directly activated via binding to a subset of BH3-only proteins known as “activators” (i.e. BCL-2 interacting mediator of cell death (BIM), BCL-2 interacting domain death agonist (BID), P53-upregulated modulator of apoptosis (PUMA), and phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1, best known as Noxa) [32][33]. The anti-apoptotic members of the BCL-2 family (e.g. BCL-2, apoptosis regulator (BCL-2), BCL2-like 1 (best known as BCL-XL) and Myeloid Leukemia Sequence 1 (MCL-1)) can also bind to BAX and prevent its function. BH3-only proteins known as “sensitizers” (e.g. BCL2 associated agonist of cell death (BAD), and BCL-2 modifying factor (BMF)) inactivate the anti-apoptotic BCL-2 family proteins, and therefore may also be needed for efficient BAX and BAK activation [24][32][34]. Early studies examining the interaction between BH3-only proteins and the anti-apoptotic proteins revealed differential binding patterns that have been exploited to examine the apoptotic vulnerability of various cells of interest [35][36][37][38]. Mitochondrial outer permeabilization induced by BAX and BAK, then results in the release of cytochrome c, oligomerization of the Apoptotic Protease Activating Factor-1 (APAF-1) and the activation of the executioners of apoptosis, a family of cysteine proteases termed caspases [39][40][41]. Caspases cleave intracellular targets [42], resulting in the morphological changes previously described, and cellular demise [24][43].
–
In contrast to differentiated cells, BAX is kept in an active state in hESCs and sequestered at the Golgi network [14][44] (Figure 3). Upon DNA damage, active BAX rapidly translocates from the Golgi to mitochondria, triggering apoptosis. Such a rapid response may be critical for preventing the propagation of aberrant cells in the developing embryo, or the emergence of a cancerous cell in the tissues of an adult organism, acting as sort of “cocked gun”. This “cocked gun” is disarmed once the cell differentiates [14][44], implying that the cell uses specific mechanisms for inactivation, which are unknown. An additional study followed these observations and determined that mitochondrial priming also contributes to the increased apoptosis sensitivity of hESCs compared to differentiated cells [15]. The balance between pro and anti-apoptotic factors shifts more towards the apoptotic threshold in hESCs [15][44]. How the high mitochondrial priming state is initiated and maintained in hPSCs, as well as how it is reset early during differentiation is not known. Considering the phenomenon of mitochondrial priming and the fact that BAX is constitutively activated in hPSCs, we hypothesized that proteins of the BCL-2 family have uncharacterized functions in hPSCs that are unrelated to cell death.
–
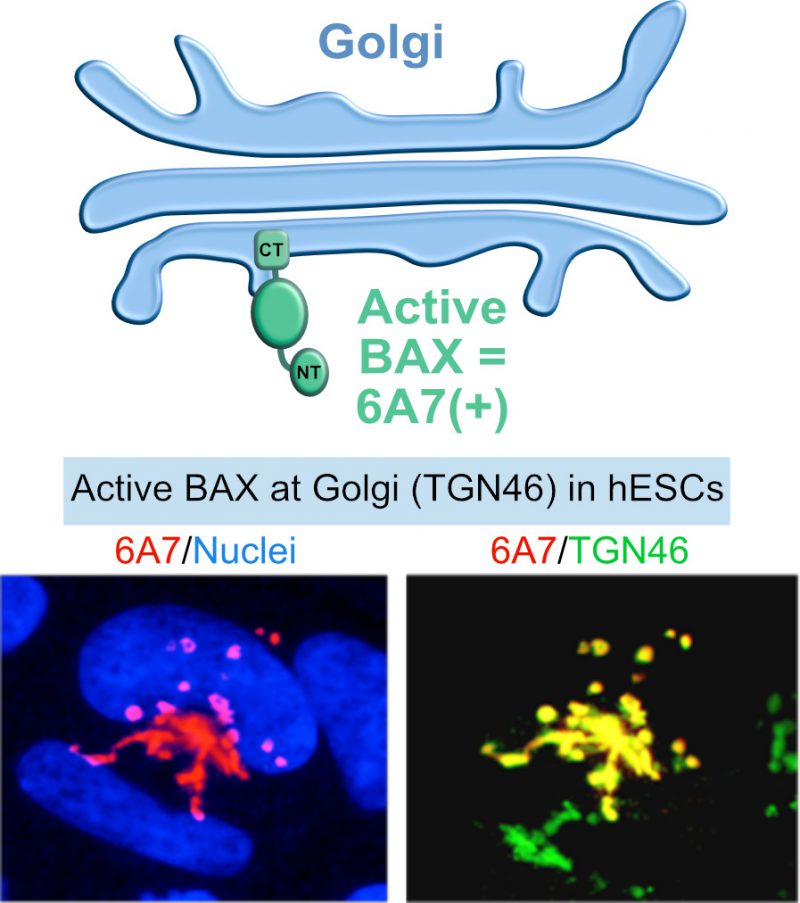 | FIGURE 3: BAX activation in stem cells. Besides high mitochondrial priming, human ESCs also have an active form of BAX at the Golgi that rapidly translocates to the mitochondria under apoptosis stimuli. Active form of BAX: BAX 6A7; Golgi apparatus marker: TGN46. |
–
Interestingly, both caspases and P53 play a major part in the generation of iPSCs. However, the mechanistic details of their involvement in somatic cell reprogramming are unknown. Apoptosis serves a defensive role by eliminating genetically abnormal and damaged cells from the pluripotent population [45][46][47][48]. Thus, antagonizing apoptosis by overexpression of anti-apoptotic BCL-2 or via repression of P53 significantly improves the efficiency of iPSC generation [49][50]. Previous studies showed that Caspase-8 deficient animals die in their early developmental stages due to improper differentiation of neural precursors [51]. This correlation between caspases and the ability of embryonic stem cells to self-renew or differentiate was later demonstrated by the report that caspase-3 and caspase-9 mutant mouse ESCs (mESCs) are unable to differentiate [52]. A caspase-dependent degradation site in the stem cell factor NANOG is conserved between mice and human protein homologues, providing insight into the mechanism by which caspases regulate reprogramming efficiency [52]. Caspase-3 dependent degradation of NANOG was reported as a key factor leading to the differentiation of hESCs and iPSCs in the absence of basic fibroblast growth factor [53]. In contrast to their role in ESCs, activated caspase-3 and caspase-8 via degradation of Retinoblastoma protein (RB), help in the de-differentiation of human fibroblasts into iPSCs [54]. Thus, caspases play a dual role by driving dedifferentiation of somatic stem cells and differentiation of ESCs, in the context of cellular and substrate availability. The use of caspase inhibitors may work as a fine regulator for differentiating ESCs/iPSCs and for reprogramming iPSCs. Hence, further investigation of underlying molecular mechanisms of caspase regulation is needed.
–
Numerous studies have demonstrated that P53 is essential for embryonic development and cellular differentiation [55]. P53 transcriptional activity in cellular differentiation is context dependent having contradictory results in various cell types [55]. For instance, P53 enhances the differentiation of both murine and human ESCs in the presence of retinoic acid and DNA damage [56][57] whereas, P53 is also shown to promote hematopoietic stem cell (HSC) quiescence and inhibit neural stem cell proliferation [58][59]. Use of the MDM2 inhibitor nutlin-3a results in rapid accumulation of P53 and subsequent induction of apoptosis and rapid differentiation in hESCs, but it has a minimal effect in somatic cells [60][61]. Similar to the observations that stabilization of P53 plays a role in differentiation of ESCs, various studies have shown that inactivation of P53 helps in reprogramming of somatic cells to iPSCs [49][62][63]. Considering that iPSCs and malignant somatic cells share similar characteristics (higher proliferation, transcriptional and metabolic status), the negative role of P53 in cellular reprogramming is not unexpected. Mechanistic insights for P53's inhibitory role in cellular reprogramming involves P53 mediated targeting of miR-34a that suppresses the expression of Sox2, MYC and NANOG [64], regulation of CDKN1A that attenuates cell division [48] and inhibition of specific epithelial genes that further suppress mesenchymal to epithelial transition [46].
–
Reprogramming efficiency of somatic cells to iPSCs is reduced in the absence of c-MYC. However, the tendency of MYC to cause apoptosis also represents a roadblock to reprogramming. A recent study indicates that reprogramming of mouse embryonic fibroblasts (MEFs) is enhanced in the absence of BAK and BAX under OKSM conditions [65]. These studies demonstrate that mitochondrial apoptosis imposes a strong MYC-dependent block to somatic cell reprogramming [10][50][65][66]. Hence, although MYC expression favors reprogramming by regulating cellular processes (transcription, translation, ATP generation) [67][68], its tendency to sensitize cells to mitochondrial apoptosis is an interesting paradox. Intriguingly, high γ-H2A.X deposition has been described in hPSCs and it has been linked to the global chromatin de-condensation required by pluripotent stem cells in order to dynamically activate transcriptional programs in response to environmental cues [69]. How these epigenetic changes are linked to increased mitochondrial apoptosis is not known. Understanding the exact mechanisms by which reprogramming induces epigenetic modifications, constitutive apoptosis sensitization and priming of pluripotent stem cells is an area in need of more investigations.
INCREASED MITOCHONDRIAL FISSION
Mitochondrial movement and fragmentation were first observed almost 100 years ago [70]. For decades these observations remained unexplored and the idea of mitochondria as isolated and static “jelly-bean” structures that serve as “power houses” of the cells became an unrefuted dogma. The development of new technologies made it easier to track mitochondria in live cells. These studies revealed the remarkable ability of mitochondria to move and to continually divide and fuse [71][72][73]. Further studies have demonstrated that mitochondrial dynamics are crucial for normal physiology from yeast to mammals. Emerging studies demonstrate a connection between mitochondrial dynamics machinery, apoptosis and mitophagy. In addition, several human diseases result from mutations in fusion and fission proteins further highlighting the relevance of mitochondrial fission and fusion events in cellular homeostasis [8][74][75][76][77] .
–
Interestingly, as differentiated cells transition to a stem cell state, the mitochondrial network undergoes dramatic changes. Mitochondria of stem cells contain functionally immature mitochondria with a globular shape, poorly developed cristae, and perinuclear localization, an indicator of a less active mitochondrial state [78][79], whereas differentiated cells, like fibroblasts, have a complex morphology with well-developed cristae, dense matrix, and elongated appearance. It is not completely understood how increased mitochondrial fragmentation is maintained in stem cells, however, constitutive activation of the dynamin-related guanosine triphosphates (GTPases) is likely a contributing factor [79].
–
Dynamin-related GTPases control mitochondrial dynamics by allowing opposing processes of division and fusion to work in concert maintaining overall shape and number of mitochondria [76][79]. Large dynamin-related GTPase proteins (DRPs) are highly conserved and have the ability to self-assemble and hydrolyze GTP to control mitochondrial fusion and fission [80][81]. In mammals, fission or fragmentation of the mitochondrial network is mediated by dynamin related protein-1 (DRP-1). DRP-1 is primarily localized in the cytosol and recruited to mitochondria during fission. Activation of DRP-1 is mediated by post translational modifications including phosphorylation, sumoylation, O-GlcNAcylation and ubiquitination which are thought to enhance its recruitment to mitochondrial receptors [83]. The impact of these post-translational modifications for maintaining the homeostatic mitochondrial fragmentation in pluripotent stem cells is not known. Modified DRP-1 oligomerizes around mitochondria and constricts the network severing both mitochondrial inner and outer membranes. The structural domains and mechanistic details of action for DRP1 have been clearly elucidated [8][80][81][82][83][84][85][86]. DRP-1 is also involved in promoting mitochondrial fragmentation during apoptosis [87] (reviewed in next section).
–
Fusion of mitochondria requires Mitofusin 1 (MFN1), Mitofusin 2 (MFN2), and Optic Atrophy 1 (OPA1) to fuse the outer and inner mitochondrial membranes [88][89][90]. Fusion is key for mitochondrial DNA (mtDNA) homogenization and assembly of electron transport chain. MFN1 and MFN2 are anchored in the outer mitochondrial membrane and allow for formation of homo- or hetero-dimers with MFNs facilitating fusion. The inner membrane is fused afterwards in a similar fashion by OPA1. OPA1 is localized to the inner mitochondrial membrane with GTPase domain exposed to inner membrane space. Overexpression of OPA1 leads to fragmentation of mitochondria, however, effects on cell death are not clear [91]. The mechanisms by which the activity of the dynamin-related proteins involved in fusion is regulated are not well understood, but data from in vitro assays illustrates that inner and outer mitochondrial fusion are separable and mechanistically different [92][93]. Fission is necessary for cell division and for mitophagy when damaged mitochondria have to be segregated. As mentioned previously, mitochondrial dynamics are crucial for normal physiology from yeast to mammals [94]. Imbalance in the process of fusion and fission leads to severe pathophysiological conditions. These range from the inability to survive past mid-gestation in MFN1, MFN2, OPA1, or DRP-1 deficient mice [90][90][95][96][97], to neurodegenerative diseases such as Charcot-Marie-Tooth syndrome and dominant optic atrophy [88][89][98][99] caused by mutations in MFN2 and OPA1.
–
The BCL-2 family has recently been implicated as a key factor in maintaining stem cell self-renewal and pluripotency. Inhibition of pro-apoptotic BAX and BAK proteins has been reported to be required for mitochondrial fusion [80][100][101][102]. BAX has been suggested to regulate fusion by interacting with MFN1 and/or MFN2 [102][103]. BCL-xL, an anti-apoptotic protein, has been shown to be highly expressed at the mitochondria of adult neurons and required for normal brain development [104]. BCL-xL appears to affect mitochondrial dynamics in mammalian neurons resulting in an increment of the length/size of mitochondria and the localization of mitochondria to synapses [105][106]. Furthermore, the anti-apoptotic protein MCL-1 appears to be involved in the regulation of mitochondrial dynamics and the maintenance of pluripotency [10]. MCL-1 appears to interact with DRP-1 and OPA1 in hPSCs, and potentially other BCL-2 family members. This interaction may be critical for the modulation of mitochondrial dynamics (Figure 4). A recent study further demonstrates that the BH3-only protein BID also regulates mitochondrial morphology and cristae organization [12]. The functional implication of a potential MCL-1 and BID interaction in maintaining pluripotency and self-renewal ability of hPSCs has not yet been explored. Revealing the mechanistic link between the mitochondrial dynamics machinery and the BCL-2 family represents a unique opportunity for increasing our understanding of how these mitochondrial signaling pathways interact to regulate cell fate.
–
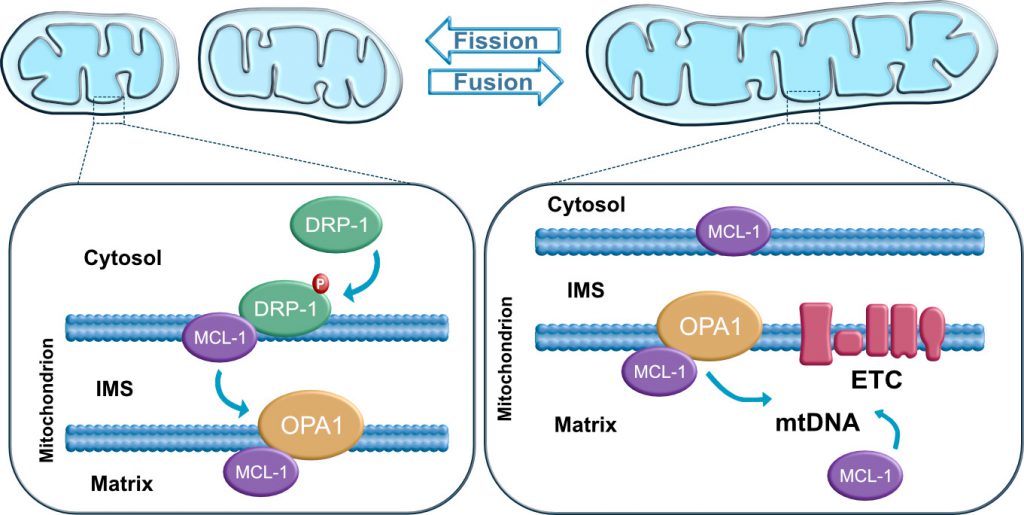 | FIGURE 4: Mitochondrial dynamics. Mitochondrial fusion and fission are regulated by guanosine triphosphatases (GTPases) proteins: DRP1 mediates fission, OPA1 and Mitofusins (not shown) regulate mitochondrial fusion. In stem cells, the anti-apoptotic protein MCL1 has been shown to interact with DRP1 at the outer mitochondrial membrane and with OPA1 at the matrix. |
–
Mitochondrial remodeling during apoptosis
The mitochondrial pathway of apoptosis causes the remodeling of mitochondrial structure that ultimately enables the release of cytochrome c, the defining moment of apoptosis [87][101][107][108][109][110][111][112]. Some studies suggest that BAX/BAK-mediated outer mitochondrial membrane permeabilization is not sufficient for cytochrome c release during apoptosis, but rather it requires mitochondrial fragmentation to occur first. Activation of BAX and BAK may lead to changes in mitochondrial cristae structure mediated by OPA1 monomerization which drives remodeling and opening of cristae junctions [113][114]. It is clear that the fragmentation of the mitochondria during apoptosis is independent of caspase activity [115], and it takes place through two coordinated, but independent, events: opening of cristae junctions, where cytochrome c is bound, and formation of the outer membrane pores [87][111][116][117][118][119][120].
–
DRP-1 colocalizes with the BAX/BAK pores [107][121][122] where it promotes disintegration of the mitochondrial network. The fragmented mitochondria collapse in a perinuclear pattern and show decreased and non-directed motility. Consistent with the increased mitochondrial fragmentation, mitochondrial fusion has also been shown to be blocked once apoptosis is activated [123].
–
Endoplasmic reticulum (ER) tubules frequently cross paths with mitochondria at points of impending fission and mark sites of mitochondrial division, a phenomenon known as ER-associated mitochondrial division (ERMD) [75][124]. These studies also indicate that the ER might play an active role during the early stages of fission, even before DRP-1 severs the mitochondria. At these “hotspots” mitochondria are constricted and allow for assembly of the diversion DRP helix. The ER may be able to alter mitochondrial membrane composition, facilitate factors, such as MFF, on the inside and/or outside of mitochondria to promote fission; however, the mechanisms underlying ER-mitochondrial microdomain or ER-mitochondrial constriction is not well established [75]. While the function of DRP1 in constricting the mitochondria has been established using cancer cell lines, the exact mechanism by which DRP-1 regulates fragmentation during apoptosis or by which it maintains the constitutive fragmentation of the mitochondrial network in stem cells and some cancer stem cells is less clear. It will be interesting to determine if the DRP-1 mechanism of action and function in homeostatic conditions and stressed conditions is modified in stem cells and cancer stem cells where mitochondria are maintained in a more fragmented state.
–
While the high mitochondrial priming described in the first section and the increased mitochondrial fragmentation are two fundamental features that accompany entry into the pluripotent state, the protein network and exact signaling events that are modulating these changes remain elusive. We speculate that the BCL-2 family mediates both of these properties, and that reprogramming of differentiated cells into stem cells provides a useful tool to dissect this intriguing possibility. Examining the connection between mitochondrial priming, fragmentation and the acquisition of a particular stem cell fate opens an exciting opportunity for future studies to determine whether these properties are mechanistically related.
DECREASED MITOCHONDRIAL-DEPENDENT METABOLISM
The increased sensitivity to cell death as well as the structural and functional remodeling of the mitochondrial network described in previous sections are accompanied by an essential switch from oxidative phosphorylation (OXPHOS) to glycolysis during reprogramming. The detailed molecular mechanisms and the temporal regulation underlying this switch remains unclear. ESCs and iPSCs have a similar metabolic profile to many cancer cells that predominantly depend on glycolysis for rapid proliferation and self-renewal. There are landmark papers on the characterization of the differences in metabolic profiles between various stages of pluripotency in mPSCs (i.e., naïve PSCs, primed PSCs and epiblast stem cells) [125][126][127]. In this section we will describe the main metabolic pathways involved in promoting cellular reprogramming and maintaining pluripotency in human PSCs [128].
–
Oxidative phosphorylation (OXPHOS)
OXPHOS is a primary source of ATP production in eukaryotic cells. A series of enzymes residing in the inner mitochondrial membrane oxidize the products of glycolysis and citric acid cycle to release protons into the inter-membrane space in the presence of oxygen. This establishes a proton gradient that drives the ATP synthase which shuttles back the hydrogen ions and produces 30 ATP molecules in the process, but it requires oxygen.
–
Energy production of PSCs rely heavily on glycolysis over OXPHOS [129][130][131]. While OXPHOS results in increased production reactive oxygen species (ROS) that could be deleterious, low ROS levels produced at the mitochondria have been shown to be critical for signaling and cell survival [132]. Thus, maintaining functional respiratory complexes may be critical for generating endogenous levels of ROS critical for stem cell renewal and pluripotency. Mitochondrial uncoupling protein 2 (UCP2)-mediated suppression of OXPHOS is required for the maintenance of pluripotency. UCP2 decouples glycolysis from OXPHOS by shunting pyruvate out of the mitochondria [133]. However, it is not completely understood whether this suppression of OXPHOS in PSCs still results in permissive low levels of endogenous ROS. Redox levels and pathways involved in redox signaling have not been thoroughly studied during cellular reprogramming, and remain an open area of investigation.
–
Glycolysis
Glycolysis involves ten reactions in the cytosol that rapidly catabolizes each six-carbon glucose molecule to produce two ATP molecules, without the requirement of oxygen. Glycolysis generates pyruvate, which in most cell types, can be shunted into two metabolic fates: in the presence of oxygen, pyruvate enters the mitochondria where it is oxidized to acetyl-CoA via pyruvate dehydrogenase (PDH), whereas in the absence of oxygen, pyruvate is reduced into lactate via lactate dehydrogenase (LDH) [134][135][136][137][138]. The intermediates in glycolysis can also be shunted into macromolecule synthesis during rapid cell growth. Thus, while it is a less efficient source of energy, glycolysis can generate both anabolic growth intermediates and ATP very rapidly owing to the much higher speed of glycolysis reactions. Primed pluripotent stem cells utilize glycolysis as a primary source of energy, converting glucose to lactate instead of directing the produced acetyl-CoA into the tricarboxylic acid cycle (TCA) cycle at the mitochondria as seen in mature differentiated cells which mostly rely on the TCA cycle for energy production [139][140][141]. Glycolysis in PSCs is crucial for maintaining pluripotency. In fact, conversion to glycolysis is necessary for successful reprogramming as inhibition of glycolysis in hESCs has been shown to result in apoptosis and cell cycle arrest [142].
–
Interestingly, hypoxic conditions are known to stimulate glycolysis and have been shown to prevent the spontaneous differentiation of hESCs [143][144]. As hypoxic niches are the primary residing locations for PSCs, glycolytic metabolism might represent an adaptation to their surroundings. The increased senescence in MSCs cultured under normoxia (21% O2) compared to when cultured under 5% O2 levels supports this hypothesis [145]. Hypoxia inducible factor-1 (HIF-1) regulates transcriptional activities and induces a switch from OXPHOS to glycolysis followed by suppression of mitochondrial biogenesis in response to hypoxia and thus, represent an important link between mitochondrial metabolism in PSCs and cellular reprogramming [146][147][148][149].
–
Methionine metabolism
Human PSCs use high levels of methionine instead of threonine as seen in murine PSCs [126][150]. This is because threonine dehydrogenase, which catabolizes threonine into 2-amino-3-ketobutyrate, evolved into a pseudogene in humans, unlike most other mammals [151]. Methionine provides the methyl group for many histone methyltransferases. Uptake of methionine from the culture media is required to maintain S-adenosylmethionine (SAM) levels in hPSCs. SAM is a methyl donor for histone methyltransferases (HMT) and DNA methyltransferases (DNMTs). Methionine deprivation results in a rapid decrease in SAM, loss of H3K4me3, and reduced NANOG expression which triggers human PSCs to differentiate. Prolonged methionine deprivation leads to cell cycle arrest and apoptosis in human PSCs [150]. SAM is consumed by NNMT (nicotinamide N-methyltransferase) which has been found upregulated in naïve hPSCs [152]. Thus, appropriate levels of methionine in the culture media are required to maintain SAM levels and global DNA and histone methylation, which are important for the maintenance of pluripotency [126].
–
Recent findings by Vernardis and collaborators correlate changes in metabolism to the differential responses of hiPSCs and hESCS, after prolonged exposure to ROCK (Rho kinase) inhibitor [153]. ROCK inhibition enables maintenance of stem cell phenotype. The authors assessed the effect of ROCK inhibition on the metabolic characteristics of cells over a 96 hours (h) of culture period. Both hPSCs and hiPSCs showed downregulation of the methionine pathway following 12 h and 24 h exposure and upregulation after 48 h. This suggests that both hPSCs and hiPSCs lose their highly proliferative characteristics after exposure to ROCK inhibition and regain their proliferation potential following adaptation to the new culture conditions. Interestingly, though, no differences in the gene expression, protein levels and physiology of hESCs and hiPSCs were observed, a differential expression of metabolic regulators p53 and mTORC1 revealed the fluctuating state of metabolism. Collectively, these and other studies have revealed the importance of methionine metabolism in integrating extrinsic amino acid information with the intrinsic epigenomics and pluripotency state to determine the cell fate of PSCs [154][155][156][157].
–
Acetyl-CoA
While the importance of metabolism on SAM levels and global methylation patterns has been studied in detail, the regulation of other key metabolites, such as acetyl-CoA, in PSCs and iPSCs needs additional investigation. Acetyl-CoA is not only a substrate for TCA cycle but it may serve for histone acetylation in PSCs for maintaining their pluripotency [158]. By performing a high resolution nuclear magnetic resonance experiment, Moussaieff and collaborators identified a metabolic transition marked by loss of acetyl-CoA that leads to histone deacetylation, during hPSC differentiation [159]. The lipogenic enzyme acetyl-CoA carboxylase has been found upregulated in iPSCs and their inhibition is linked to decreased reprogramming efficiency [160]. Likewise, chemical inhibition of histone deacetylases is shown to promote reprogramming of somatic cells to iPSCs [161]. These studies suggest that it is via shunting of pyruvate-cytosolic acetyl-CoA that glycolysis may contribute to pluripotency regulation [162].
–
A recent study reported that cytosolic acetyl-CoA, whichis produced through glycolysis and the pyruvate-derived citrate flux via ATP citrate lyase (ACLY) is inhibited during PSC differentiation [159]. Acetyl-CoA blocks histone deacetylation and stem cell differentiation, while acetate, an alternative precursor of cytosolic acetyl-CoA, delays PSC differentiation by promoting histone acetylation in a dose-dependent fashion [159].Thus, a glycolytic switch that regulates histone acetylation can have a profound effect on the ability of stem cells to differentiate [159]. Of note, the decrease in OXPHOS might be causal for the activation of glycolysis during reprogramming, as (OXPHOS-derived) ATP is a potent allosteric inhibitor of a number of enzymes involved in glycolysis [163]. In sum, glycolysis may remodel the metabolome and facilitate the reprogramming of somatic cells into iPSCs [129][164][165].
–
While it is conventionally thought that metabolism is altered as a consequence of the chosen cell fate (i.e., metabolic demand drives the cell's metabolic program), an intriguing possibility is that metabolism itself can dictate stem cell renewal and/or differentiation by altering transcriptional networks that modulate cell fate [134]. It will be interesting to determine when during reprogramming and differentiation these metabolic changes take place. Characterization of the temporal regulation of metabolism, mitochondrial dynamics, and apoptosis during stem cell fate decisions could provide critical insight into if the changes in these processes are a result of the stem cell state, or a necessary factor for maintenance of the state of the cell.
CONCLUDING REMARKS
Mitochondria are a crucial source for fuel and intermediate metabolites that are essential for many cellular functions. These organelles house key proteins involved in the regulation of apoptosis and metabolism while maintaining a highly dynamic network that is essential for their function. Decades of studying these organelles in isolation have allowed for the elucidation of the main components involved in apoptosis, OXPHOS and mitochondrial dynamics, but we know less about the crosstalk between these pathways and how they are regulated as mitochondria communicate within themselves and with other cellular membranes. Many questions about mechanisms and integration of these mitochondrial changes and signaling networks remain unanswered. Future studies should be aimed to understand how these pathways intersect to regulate cell survival, what factors control the switch in mitochondrial network status, turnover, and connectivity to the ER as cells undergo reprogramming; how do changes in metabolic state and metabolite levels affect epigenetic enzymes that regulate gene expression; and how is respiratory capacity increased with differentiation and lowered with reprogramming. Answering these questions could lead to a complete understanding of how mitochondrial biology and function modulate cell fate.
–
Recent advances in the field of stem cell biology and in cellular reprogramming technology in particular, have created new opportunities in understanding human disease, drug discovery, and regenerative medicine. The generation of iPSCs reveals the remarkable plasticity associated with differentiated cells and provides unprecedented opportunity to model diseases using patient samples. In addition to transcriptional and epigenetic remodeling, cellular reprogramming is also accompanied by dramatic changes in the structure and function of the mitochondria. In this review, we describe three main mitochondrial events linked with reprogramming: increase of cell death sensitivity, fission of mitochondrial network, and decreased OXPHOS. We discussed potential mechanisms underlying the rewiring of the cellular state. It is clear, that we are just beginning to understand the integral roles played by mitochondrial morphology and function in development and diseases. There are also other areas of exciting opportunities. Today, it is possible to culture additional types of human pluripotent stem cells that model cells from preimplantation and post-implantation embryos [125][166]. These different phases of pluripotency, for example, naïve state (which resembles the pre-implantation blastocyst inner cell mass), primed state (which resemble the post-implantation epiblast cells) and extended PSCs (which can generate embryonic and extraembryonic tissues), can all now be cultured in vitro simply by adapting different cell culture parameters. This possibility opens the potential to examine the function of mitochondrial morphology, dynamics and function in the context of the developmental potential of all of these model systems.
–
Pioneer work from Ruth Slack's group [11] highlighted the physiological significance of mitochondrial dynamics for stem cell identity and fate during mouse brain development. Studies in human brain have been limited by the lack of model systems. The emergence of three-dimensional models, known as organoids [167][168][169][170], has opened a new avenue of discovery and could provide an additional model system to study the effects of disrupting mitochondrial morphology/function on early human brain development. While human stem cells provide many advantages, it is difficult to model many stages of maturation, such as neurodevelopmental processes of myelination, gliogenesis etc., which may not occur in the time that a monolayer culture can be maintained. Understanding the molecular events governing the mitochondria-related hallmarks described in this review, in the context of three-dimensional tissues could reveal new areas of discovery on the impact of disrupting mitochondrial function in the development of degenerative diseases. Organoids better recapitulate functional, structural, and architectural complexity of organs and their cellular diversity and thus, provide a powerful tool to dissect the principles of mitochondrial biology we have learned from other systems.
–
Recent discoveries have evidenced the interplay between mitochondrial dynamics, intermediate metabolism and epigenetics, thus, the traditional views of mitochondria as isolated organelles and of metabolism as a developmental byproduct have largely been refuted. To what extent mitochondrial morphology and cellular metabolism are directly wired to cellular transitions associated with development and reprograming warrants future investigations. Recent studies shed light into the essential requirement for a fine-tuned regulation of mitochondrial morphology and function for the maintenance of stem cell properties. New studies in these areas could have farreaching implications facilitating innovations leading to new treatments for patients affected by neurodevelopmental and neuropsychiatric disorders.
REFERENCES
- Liu X, Kim CN, Yang J, Jemmerson R, and Wang X (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86(1): 147–157. doi: 10.1016/s0092- 8674(00)80085-9
- Vaux DL, Cory S, and Adams JM (1988). Bcl-2 gene promotes hae- mopoietic cell survival and cooperates with c-myc to immortalize pre- B cells. Nature 335(6189): 440–442. doi: 10.1038/335440a0
- Mishra P, and Chan DC (2016). Metabolic regulation of mitochon- drial dynamics. J Cell Biol 212(4): 379–387. doi: 10.1083/jcb.201511036
- Mishra P, and Chan DC (2014). Mitochondrial dynamics and inher- itance during cell division, development and disease. Nat Rev Mol Cell Biol 15(10): 634–646. doi: 10.1038/nrm3877
- Chauhan A, Vera J, and Wolkenhauer O (2014). The systems biology of mitochondrial fission and fusion and implications for disease and aging. Biogerontology 15(1): 1–12. doi: 10.1007/s10522-013-9474-z
- Burté F, Carelli V, Chinnery PF, and Yu-Wai-Man P (2015). Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11(1): 11–24. doi: 10.1038/nrneurol.2014.228
- Suen D-F, Norris KL, and Youle RJ (2008). Mitochondrial dynamics and apoptosis. Genes Dev 22(12): 1577–1590. doi: 10.1101/gad.1658508
- Youle RJ, and van der Bliek AM (2012). Mitochondrial fission, fusion, and stress. Science 337(6098): 1062–1065. doi: 10.1126/science.1219855
- Elkholi R, Floros KV, and Chipuk JE (2011). The Role of BH3-Only Proteins in Tumor Cell Development, Signaling, and Treatment. Genes Cancer 2(5): 523–537. doi: 10.1177/1947601911417177
- Rasmussen ML, Kline LA, Park KP, Ortolano NA, Romero-Morales AI, Anthony CC, Beckermann KE, and Gama V (2018). A Non-apoptotic Function of MCL-1 in Promoting Pluripotency and Modulating Mito- chondrial Dynamics in Stem Cells. Stem Cell Reports 10(3): 684–692. doi: 10.1016/j.stemcr.2018.01.005
- Khacho M, Clark A, Svoboda DS, MacLaurin JG, Lagace DC, Park DS, and Slack RS (2017). Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neuro- genesis. Hum Mol Genet 26(17): 3327–3341. doi: 10.1093/hmg/ddx217
- Salisbury-Ruf CT, Bertram CC, Vergeade A, Lark DS, Shi Q, Heber- ling ML, Fortune NL, Okoye GD, Jerome WG, Wells QS, Fessel J, Mos- lehi J, Chen H, Roberts LJ, Boutaud O, Gamazon ER, and Zinkel SS (2018). Bid maintains mitochondrial cristae structure and function and protects against cardiac disease in an integrative genomics study. Elife 7: e40907. doi: 10.7554/eLife.40907
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, and Jones JM (1998). Embryonic stem cell lines derived from human blastocysts. Science 282(5391): 1145–1147. doi: 10.1126/science.282.5391.1145
- Dumitru R, Gama V, Fagan BM, Bower JJ, Swahari V, Pevny LH, and Deshmukh M (2012). Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol Cell 46(5): 573–583. doi: 10.1016/j.molcel.2012.04.002
- Liu JC, Guan X, Ryan JA, Rivera AG, Mock C, Agrawal V, Agarwal V, Letai A, Lerou PH, and Lahav G (2013). High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell 13(4): 483–491. doi: 10.1016/j.stem.2013.07.018
- Mathieu J, and Ruohola-Baker H (2017). Metabolic remodeling during the loss and acquisition of pluripotency. Development 144(4): 541–551. doi: 10.1242/dev.128389
- Mishra P, and Chan DC (2014). Mitochondrial dynamics and inher- itance during cell division, development and disease. Nat Rev Mol Cell Biol 15(10): 634–646. doi: 10.1038/nrm3877
- Kerr JF, Wyllie AH, and Currie AR (1972). Apoptosis: a basic biolog- ical phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4): 239–257. doi: 10.1038/bjc.1972.33
- Wong RSY (2011). Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res 30: 87. doi: 10.1186/1756-9966-30- 87
- Schimmer AD (2008). Apoptosis in leukemia: from molecular pathways to targeted therapies. Best Pract Res Clin Haematol 21(1): 5–11. doi: 10.1016/j.beha.2007.11.002
- Elmore S (2007). Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4): 495–516. doi: 10.1080/01926230701320337
- Ashkenazi A, and Dixit VM (1998). Death Receptors: Signaling and Modulation. Science 281(5381): 1305–1308. doi: 10.1126/science.281.5381.1305
- Mehlen P, and Bredesen DE (2011). Dependence receptors: from basic research to drug development. Sci Signal 4(157): mr2. doi: 10.1126/scisignal.2001521
- Galluzzi L et al. (2018). Molecular mechanisms of cell death: rec- ommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25(3): 486–541. doi: 10.1038/s41418-017-0012-4
- Vousden KH, and Prives C (2009). Blinded by the Light: The Grow- ing Complexity of p53. Cell 137(3): 413–431. doi: 10.1016/j.cell.2009.04.037
- Tiwari B, Jones AE, and Abrams JM (2018). Transposons, p53 and Genome Security. Trends Genet 34(11): 846–855. doi: 10.1016/j.tig.2018.08.003
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, and Youle RJ (1997). Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 139(5): 1281–1292. doi: 10.1083/jcb.139.5.1281
- Walensky LD, and Gavathiotis E (2011). BAX unleashed: the bio- chemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem Sci 36(12): 642–652. doi: 10.1016/j.tibs.2011.08.009
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, and Green DR (2004). Direct activation of Bax by p53 me- diates mitochondrial membrane permeabilization and apoptosis. Science 303(5660): 1010–1014. doi: 10.1126/science.1092734
- Danial NN, and Korsmeyer SJ (2004). Cell death: critical control points. Cell 116(2): 205–219. doi: 10.1016/S0092-8674(04)00046-7
- Chipuk JE, and Green DR (2008). How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol 18(4): 157–164. doi: 10.1016/j.tcb.2008.01.007
- Youle RJ, and Strasser A (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9(1): 47–59. doi: 10.1038/nrm2308
- Llambi F, Moldoveanu T, Tait SWG, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, and Green DR (2011). A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell 44(4): 517–531. doi: 10.1016/j.molcel.2011.10.001
- Opferman JT, and Kothari A (2018). Anti-apoptotic BCL-2 family members in development. Cell Death Differ 25(1): 37–45. doi: 10.1038/cdd.2017.170
- Brunelle JK, and Letai A (2009). Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci 122(Pt 4): 437–441. doi: 10.1242/jcs.031682
- Montero J, Sarosiek KA, DeAngelo JD, Maertens O, Ryan J, Ercan D, Piao H, Horowitz NS, Berkowitz RS, Matulonis U, Jänne PA, Amrein PC, Cichowski K, Drapkin R, and Letai A (2015). Drug-induced death signal- ing strategy rapidly predicts cancer response to chemotherapy. Cell 160(5): 977–989. doi: 10.1016/j.cell.2015.01.042
- Montero J, and Letai A (2016). Dynamic BH3 profiling-poking can- cer cells with a stick. Mol Cell Oncol 3(3): e1040144. doi: 10.1080/23723556.2015.1040144
- Belizário JE, Alves J, Occhiucci JM, Garay-Malpartida M, and Sesso A (2007). A mechanistic view of mitochondrial death decision pores. Braz J Med Biol Res 40(8): 1011–1024. doi: 10.1590/s0100- 879×2006005000109
- Inoue S, Browne G, Melino G, and Cohen GM (2009). Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ 16(7): 1053–1061. doi: 10.1038/cdd.2009.29
- Kim H-E, Du F, Fang M, and Wang X (2005). Formation of apopto- some is initiated by cytochrome c-induced dATP hydrolysis and subse- quent nucleotide exchange on Apaf-1. Proc Natl Acad Sci USA 102(49): 17545–17550. doi: 10.1073/pnas.0507900102
- Taylor RC, Cullen SP, and Martin SJ (2008). Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9(3): 231–241. doi: 10.1038/nrm2312
- Ramirez MLG, and Salvesen GS (2018). A primer on caspase mech- anisms. Semin Cell Dev Biol 82: 79–85. doi: 10.1016/j.semcdb.2018.01.002
- Fuchs Y, and Steller H (2011). Programmed cell death in animal development and disease. Cell 147(4): 742–758. doi: 10.1016/j.cell.2011.10.033
- Gama V, and Deshmukh M (2012). Human embryonic stem cells: living on the edge. Cell Cycle 11(21): 3905–3906. doi: 10.4161/cc.22233
- Stambrook PJ, and Tichy ED (2010). Preservation of genomic integ- rity in mouse embryonic stem cells. Adv Exp Med Biol.695: 59–75. doi: 10.1007/978-1-4419-7037-4_5
- Brosh R, Assia-Alroy Y, Molchadsky A, Bornstein C, Dekel E, Madar S, Shetzer Y, Rivlin N, Goldfinger N, Sarig R, and Rotter V (2013). p53 counteracts reprogramming by inhibiting mesenchymal-to-epithelial transition. Cell Death Differ 20(2): 312–320. doi: 10.1038/cdd.2012.125
- Ohgushi M, Matsumura M, Eiraku M, Murakami K, Aramaki T, Nishiyama A, Muguruma K, Nakano T, Suga H, Ueno M, Ishizaki T, Suemori H, Narumiya S, Niwa H, and Sasai Y (2010). Molecular path- way and cell state responsible for dissociation-induced apoptosis in human pluripotent stem cells. Cell Stem Cell 7(2): 225–239. doi: 10.1016/j.stem.2010.06.018
- Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, Creyghton MP, van Oudenaarden A, and Jaenisch R (2009). Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 462(7273): 595–601. doi: 10.1038/nature08592
- Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, Okita K, and Yamanaka S (2009). Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 460(7259): 1132–1135. doi: 10.1038/nature08235
- Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, and Izpisúa Belmonte JC (2009). Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460(7259): 1140–1144. doi: 10.1038/nature08311
- Sakamaki K, Inoue T, Asano M, Sudo K, Kazama H, Sakagami J, Sakata S, Ozaki M, Nakamura S, Toyokuni S, Osumi N, Iwakura Y, and Yonehara S (2002). Ex vivo whole-embryo culture of caspase-8- deficient embryos normalize their aberrant phenotypes in the devel- oping neural tube and heart. Cell Death Differ 9(11): 1196–1206. doi: 10.1038/sj.cdd.4401090
- Fujita J, Crane AM, Souza MK, Dejosez M, Kyba M, Flavell RA, Thomson JA, and Zwaka TP (2008). Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell 2(6): 595–601. doi: 10.1016/j.stem.2008.04.001
- Wang X, Lin G, Martins-Taylor K, Zeng H, and Xu R-H (2009). Inhibi- tion of caspase-mediated anoikis is critical for basic fibroblast growth factor-sustained culture of human pluripotent stem cells. J Biol Chem 284(49): 34054–34064. doi: 10.1074/jbc.M109.052290
- Li F, He Z, Shen J, Huang Q, Li W, Liu X, He Y, Wolf F, and Li C-Y (2010). Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell 7(4): 508–520. doi: 10.1016/j.stem.2010.09.003
- Jain AK, and Barton MC (2018). p53: emerging roles in stem cells, development and beyond. Development 145(8): dev158360. doi: 10.1242/dev.158360
- Akdemir KC, Jain AK, Allton K, Aronow B, Xu X, Cooney AJ, Li W, and Barton MC (2014). Genome-wide profiling reveals stimulus- specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res 42(1): 205–223. doi: 10.1093/nar/gkt866
- Li M, He Y, Dubois W, Wu X, Shi J, and Huang J (2012). Distinct regulatory mechanisms and functions for p53-activated and p53- repressed DNA damage response genes in embryonic stem cells. Mol Cell 46(1): 30–42. doi: 10.1016/j.molcel.2012.01.020
- Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A, and Nimer SD (2009). p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4(1): 37–48. doi: 10.1016/j.stem.2008.11.006
- Meletis K, Wirta V, Hede S-M, Nistér M, Lundeberg J, and Frisén J (2006). p53 suppresses the self-renewal of adult neural stem cells. Development 133(2): 363–369. doi: 10.1242/dev.02208
- Setoguchi K, TeSlaa T, Koehler CM, and Teitell MA (2016). P53 Regulates Rapid Apoptosis in Human Pluripotent Stem Cells. J Mol Biol 428(7): 1465–1475. doi: 10.1016/j.jmb.2015.07.019
- Maimets T, Neganova I, Armstrong L, and Lako M (2008). Activa- tion of p53 by nutlin leads to rapid differentiation of human embryon- ic stem cells. Oncogene 27(40): 5277–5287. doi: 10.1038/onc.2008.166
- Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, Khalil A, Rheinwald JG, and Hochedlinger K (2009). Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460(7259): 1145–1148. doi: 10.1038/nature08285
- Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez- Capetillo O, Serrano M, and Blasco MA (2009). A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460(7259): 1149–1153. doi: 10.1038/nature08287
- Choi YJ, Lin C-P, Ho JJ, He X, Okada N, Bu P, Zhong Y, Kim SY, Ben- nett MJ, Chen C, Ozturk A, Hicks GG, Hannon GJ, and He L (2011). miR- 34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol 13(11): 1353–1360. doi: 10.1038/ncb2366
- Kim EJY, Anko M-L, Flensberg C, Majewski IJ, Geng F-S, Firas J, Huang DCS, van Delft MF, and Heath JK (2018). BAK/BAX-Mediated Apoptosis Is a Myc-Induced Roadblock to Reprogramming. Stem Cell Reports 10(2): 331–338. doi: 10.1016/j.stemcr.2017.12.019
- Li Y, Feng H, Gu H, Lewis DW, Yuan Y, Zhang L, Yu H, Zhang P, Cheng H, Miao W, Yuan W, Cheng S-Y, Gollin SM, and Cheng T (2013). The p53-PUMA axis suppresses iPSC generation. Nat Commun 4: 2174. doi: 10.1038/ncomms3174
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, Zhao K, and Levens D (2012). c-Myc is a uni- versal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151(1): 68–79. doi: 10.1016/j.cell.2012.08.033
- Polo JM, Anderssen E, Walsh RM, Schwarz BA, Nefzger CM, Lim SM, Borkent M, Apostolou E, Alaei S, Cloutier J, Bar-Nur O, Cheloufi S, Stadtfeld M, Figueroa ME, Robinton D, Natesan S, Melnick A, Zhu J, Ramaswamy S, and Hochedlinger K (2012). A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 151(7): 1617–1632. doi: 10.1016/j.cell.2012.11.039
- Banáth JP, Bañuelos CA, Klokov D, MacPhail SM, Lansdorp PM, and Olive PL (2009). Explanation for excessive DNA single-strand breaks and endogenous repair foci in pluripotent mouse embryonic stem cells. Exp Cell Res 315(8): 1505–1520. doi: 10.1016/j.yexcr.2008.12.007
- Lewis MR, and Lewis WH (1914). Mitochondria in Tissue Culture. Science 39(1000): 330–333. doi: 10.1126/science.39.1000.330
- Johnson LV, Walsh ML, Bockus BJ, and Chen LB (1981). Monitoring of relative mitochondrial membrane potential in living cells by fluores- cence microscopy. J Cell Biol 88(3): 526–535. doi: 10.1083/jcb.88.3.526
- Bereiter-Hahn J, and Vöth M (1994). Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mito- chondria. Microsc Res Tech 27(3): 198–219. doi: 10.1002/jemt.1070270303
- Rizzuto R, Brini M, De Giorgi F, Rossi R, Heim R, Tsien RY, and Poz- zan T (1996). Double labelling of subcellular structures with organelle- targeted GFP mutants in vivo. Curr Biol 6(2): 183–188. doi: 10.1016/s0960-9822(02)00451-7
- Westermann B (2010). Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11(12): 872–884. doi: 10.1038/nrm3013
- Friedman JR, and Nunnari J (2014). Mitochondrial form and func- tion. Nature 505(7483): 335–343. doi: 10.1038/nature12985
- Nunnari J, and Suomalainen A (2012). Mitochondria: In Sickness and in Health. Cell 148(6): 1145–1159. doi: 10.1016/j.cell.2012.02.035
- Chan DC (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46: 265–287. doi: 10.1146/annurev-genet-110410-132529
- Facucho-Oliveira JM, and St John JC (2009). The relationship be- tween pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev 5(2): 140–158. doi: 10.1007/s12015-009-9058-0
- Chung S, Arrell DK, Faustino RS, Terzic A, and Dzeja PP (2010). Glycolytic network restructuring integral to the energetics of embry- onic stem cell cardiac differentiation. J Mol Cell Cardiol 48(4): 725– 734. doi: 10.1016/j.yjmcc.2009.12.014
- Hoppins S, Lackner L, and Nunnari J (2007). The machines that divide and fuse mitochondria. Annu Rev Biochem 76: 751–780. doi: 10.1146/annurev.biochem.76.071905.090048
- Praefcke GJK, and McMahon HT (2004). The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol 5(2): 133–147. doi: 10.1038/nrm1313
- Meglei G, and McQuibban GA (2009). The dynamin-related protein Mgm1p assembles into oligomers and hydrolyzes GTP to function in mitochondrial membrane fusion. Biochemistry 48(8): 1774–1784. doi: 10.1021/bi801723d
- van der Bliek AM, and Payne GS (2010). Dynamin subunit interac- tions revealed. Dev Cell 18(5): 687–688. doi: 10.1016/j.devcel.2010.05.001
- Chappie JS, Acharya S, Leonard M, Schmid SL, and Dyda F (2010). G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature 465(7297): 435–440. doi: 10.1038/nature09032
- Gao S, von der Malsburg A, Paeschke S, Behlke J, Haller O, Kochs G, and Daumke O (2010). Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 465(7297): 502–506. doi: 10.1038/nature08972
- Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, and Hinshaw JE (2011). Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol 18(1): 20–26. doi: 10.1038/nsmb.1949
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, and Youle RJ (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1(4): 515–525. doi: 10.1016/s1534-5807(01)00055-7
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, and Wissinger B (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26(2): 211–215. doi: 10.1038/79944
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, and Hamel CP (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related pro- tein, is mutated in dominant optic atrophy. Nat Genet 26(2): 207–210. doi: 10.1038/79936
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, and Chan DC (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondri- al fusion and are essential for embryonic development. J Cell Biol 160(2): 189–200. doi: 10.1083/jcb.200211046
- Misaka T, Miyashita T, and Kubo Y (2002). Primary structure of a dynamin-related mouse mitochondrial GTPase and its distribution in brain, subcellular localization, and effect on mitochondrial morpholo- gy. J Biol Chem 277(18): 15834–15842. doi: 10.1074/jbc.M109260200
- Legros F, Lombès A, Frachon P, and Rojo M (2002). Mitochondrial fusion in human cells is efficient, requires the inner membrane poten- tial, and is mediated by mitofusins. Mol Biol Cell 13(12): 4343–4354. doi: 10.1091/mbc.e02-06-0330
- Meeusen S, McCaffery JM, and Nunnari J (2004). Mitochondrial fusion intermediates revealed in vitro. Science 305(5691): 1747–1752. doi: 10.1126/science.1100612
- Labbé C, Rayaprolu S, Soto-Ortolaza A, Ogaki K, Uitti RJ, Wszolek ZK, and Ross OA (2014). Investigating FUS variation in Parkinson’s disease. Parkinsonism Relat Disord 20 (Suppl 1): S147-149. doi: 10.1016/S1353-8020(13)70035-X
- Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, Nicols PP, Boulton ME, and Votruba M (2007). Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mito- chondrial morphology, optic nerve structure and visual function. Hum Mol Genet 16(11): 1307–1318. doi: 10.1093/hmg/ddm079
- Alavi MV, Bette S, Schimpf S, Schuettauf F, Schraermeyer U, Wehrl HF, Ruttiger L, Beck SC, Tonagel F, Pichler BJ, Knipper M, Peters T, Laufs J, and Wissinger B (2007). A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 130(Pt 4): 1029–1042. doi: 10.1093/brain/awm005
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y-I, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, and Mihara K (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and syn- apse formation in mice. Nat Cell Biol 11(8): 958–966. doi: 10.1038/ncb1907
- Waterham HR, Koster J, van Roermund CWT, Mooyer PAW, Wan- ders RJA, and Leonard JV (2007). A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 356(17): 1736–1741. doi: 10.1056/NEJMoa064436
- Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quat- trone A, Battaloglu E, Polyakov AV, Timmerman V, Schröder JM, Vance JM, and Battologlu E (2004). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36(5): 449–451. doi: 10.1038/ng1341
- Cleland J, Gillani R, Bienen EJ, and Sadosky A (2011). Assessing dimensionality and responsiveness of outcomes measures for patients with low back pain. Pain Pract 11(1): 57–69. doi: 10.1111/j.1533- 2500.2010.00390.x
- Karbowski M, Norris KL, Cleland MM, Jeong S-Y, and Youle RJ (2006). Role of Bax and Bak in mitochondrial morphogenesis. Nature 443(7112): 658–662. doi: 10.1038/nature05111
- Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ, and Nunnari J (2011). The soluble form of Bax regulates mitochon- drial fusion via MFN2 homotypic complexes. Mol Cell 41(2): 150–160. doi: 10.1016/j.molcel.2010.11.030
- Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie Z-J, and Dong Z (2007). Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA 104(28): 11649–11654. doi: 10.1073/pnas.0703976104
- Chen Y-B, Aon MA, Hsu Y-T, Soane L, Teng X, McCaffery JM, Cheng W-C, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, O’Rourke B, Ko YH, Pedersen PL, Kaczmarek LK, Jonas EA, and Hardwick JM (2011). Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol 195(2): 263–276. doi: 10.1083/jcb.201108059
- Berman SB, Pineda FJ, and Hardwick JM (2008). Mitochondrial fission and fusion dynamics: the long and short of it. Cell Death Differ 15(7): 1147–1152. doi: 10.1038/cdd.2008.57
- Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, Bossy-Wetzel E, Bennett MVL, Pypaert M, Hickman JA, Smith PJS, Hardwick JM, and Jonas EA (2008). Bcl-xL induces Drp1-dependent synapse formation in cultured hippo- campal neurons. Proc Natl Acad Sci USA 105(6): 2169–2174. doi: 10.1073/pnas.0711647105
- Karbowski M, Lee Y-J, Gaume B, Jeong S-Y, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, and Youle RJ (2002). Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol 159(6): 931–938. doi: 10.1083/jcb.200209124
- Kushnareva Y, Andreyev AY, Kuwana T, and Newmeyer DD (2012). Bax activation initiates the assembly of a multimeric catalyst that facilitates Bax pore formation in mitochondrial outer membranes. PLoS Biol 10(9): e1001394. doi: 10.1371/journal.pbio.1001394
- Colell A, Ricci J-E, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A, Waterhouse NJ, Li CW, Mari B, Barbry P, Newmeyer DD, Beere HM, and Green DR (2007). GAPDH and autoph- agy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 129(5): 983–997. doi: 10.1016/j.cell.2007.03.045
- Ekert PG, Read SH, Silke J, Marsden VS, Kaufmann H, Hawkins CJ, Gerl R, Kumar S, and Vaux DL (2004). Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug- treated cells die. J Cell Biol 165(6): 835–842. doi: 10.1083/jcb.200312031
- Ow Y-LP, Green DR, Hao Z, and Mak TW (2008). Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol 9(7): 532–542. doi: 10.1038/nrm2434
- Goldstein JC, Waterhouse NJ, Juin P, Evan GI, and Green DR (2000). The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol 2(3): 156–162. doi: 10.1038/35004029
- Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, Kuwana T, Ellisman MH, and Newmeyer DD (2008). Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell 31(4): 557–569. doi: 10.1016/j.molcel.2008.07.010
- Cogliati S, Enriquez JA, and Scorrano L (2016). Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem Sci 41(3): 261–273. doi: 10.1016/j.tibs.2016.01.001
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, and Newmeyer DD (2002). Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondri- al membrane. Cell 111(3): 331–342. doi: 10.1016/s0092- 8674(02)01036-x
- Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basañez G, Meda P, and Martinou J-C (2010). Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142(6): 889–901. doi: 10.1016/j.cell.2010.08.017
- Sinibaldi F, Howes BD, Droghetti E, Polticelli F, Piro MC, Di Pierro D, Fiorucci L, Coletta M, Smulevich G, and Santucci R (2013). Role of lysines in cytochrome c-cardiolipin interaction. Biochemistry 52(26): 4578–4588. doi: 10.1021/bi400324c
- Gao W, Pu Y, Luo KQ, and Chang DC (2001). Temporal relation- ship between cytochrome c release and mitochondrial swelling during UV-induced apoptosis in living HeLa cells. J Cell Sci 114(Pt 15): 2855– 2862. PMID: 11683418
- Lee Y, Jeong S-Y, Karbowski M, Smith CL, and Youle RJ (2004). Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 15(11): 5001–5011. doi: 10.1091/mbc.e04-04-0294
- Sheridan C, Delivani P, Cullen SP, and Martin SJ (2008). Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell 31(4): 570–585. doi: 10.1016/j.molcel.2008.08.002
- Estaquier J, and Arnoult D (2007). Inhibiting Drp1-mediated mito- chondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ 14(6): 1086–1094. doi: 10.1038/sj.cdd.4402107
- Sugioka R, Shimizu S, and Tsujimoto Y (2004). Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 279(50): 52726–52734. doi: 10.1074/jbc.M408910200
- Karbowski M, Jeong S-Y, and Youle RJ (2004). Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol 166(7): 1027–1039. doi: 10.1083/jcb.200407046
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, and Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science 334(6054): 358–362. doi: 10.1126/science.1207385
- Zhang J, Zhao J, Dahan P, Lu V, Zhang C, Li H, and Teitell MA (2018). Metabolism in Pluripotent Stem Cells and Early Mammalian Development. Cell Metabolism 27(2): 332–338. doi: 10.1016/j.cmet.2018.01.008
- Teslaa T, and Teitell MA (2015). Pluripotent stem cell energy metabolism: an update. EMBO J 34(2): 138–153. doi: 10.15252/embj.201490446
- Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, and Ru- ohola-Baker H (2012). HIF1α induced switch from bivalent to exclu- sively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J 31(9): 2103–2116. doi: 10.1038/emboj.2012.71
- Xu X, Duan S, Yi F, Ocampo A, Liu G-H, and Izpisua Belmonte JC (2013). Mitochondrial Regulation in Pluripotent Stem Cells. Cell Me- tabolism 18(3): 325–332. doi: 10.1016/j.cmet.2013.06.005
- Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA, Ramalho-Santos J, Van Houten B, and Schatten G (2011). Energy me- tabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 6(6): e20914. doi: 10.1371/journal.pone.0020914
- Folmes CDL, Dzeja PP, Nelson TJ, and Terzic A (2012). Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 11(5): 596–606. doi: 10.1016/j.stem.2012.10.002
- Chen X, Gu Q, Wang X, Ma Q, Tang H, Yan X, Guo X, Yan H, Hao J, and Zeng F (2013). Directed neuronal differentiation of mouse embry- onic and induced pluripotent stem cells and their gene expression profiles. Int J Mol Med 32(1): 25–34. doi: 10.3892/ijmm.2013.1372
- Sena LA, and Chandel NS (2012). Physiological roles of mitochon- drial reactive oxygen species. Mol Cell 48(2): 158–167. doi: 10.1016/j.molcel.2012.09.025
- Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, Jung H-J, McCaffery JM, Kurland IJ, Reue K, Lee W-NP, Koehler CM, and Teitell MA (2016). UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J 35(8): 899. doi: 10.15252/embj.201694054
- Chandel NS (2015). Evolution of Mitochondria as Signaling Orga- nelles. Cell Metab 22(2): 204–206. doi: 10.1016/j.cmet.2015.05.013
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv 2(5): e1600200. doi: 10.1126/sciadv.1600200
- Chandel NS (2014). Mitochondria as signaling organelles. BMC Biol 12: 34. doi: 10.1186/1741-7007-12-34
- Picard M, Wallace DC, and Burelle Y (2016). The rise of mito- chondria in medicine. Mitochondrion 30: 105–116. doi: 10.1016/j.mito.2016.07.003
- Wallace DC (2013). Bioenergetics in human evolution and dis- ease: implications for the origins of biological complexity and the missing genetic variation of common diseases. Philos Trans R Soc Lond, B, Biol Sci 368(1622): 20120267. doi: 10.1098/rstb.2012.0267
- Gaspar JA, Doss MX, Hengstler JG, Cadenas C, Hescheler J, and Sachinidis A (2014). Unique metabolic features of stem cells, cardio- myocytes, and their progenitors. Circ Res 114(8): 1346–1360. doi: 10.1161/CIRCRESAHA.113.302021
- Shyh-Chang N, Daley GQ, and Cantley LC (2013). Stem cell me- tabolism in tissue development and aging. Development 140(12): 2535–2547. doi: 10.1242/dev.091777
- Shyh-Chang N, and Ng H-H (2017). The metabolic programming of stem cells. Genes Dev 31(4): 336–346. doi: 10.1101/gad.293167.116
- Burgess RJ, Agathocleous M, and Morrison SJ (2014). Metabolic regulation of stem cell function. J Intern Med 276(1): 12–24. doi: 10.1111/joim.12247
- Ezashi T, Das P, and Roberts RM (2005). Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA 102(13): 4783–4788. doi: 10.1073/pnas.0501283102
- Prasad SM, Czepiel M, Cetinkaya C, Smigielska K, Weli SC, Lysdahl H, Gabrielsen A, Petersen K, Ehlers N, Fink T, Minger SL, and Zachar V (2009). Continuous hypoxic culturing maintains activation of Notch and allows long-term propagation of human embryonic stem cells without spontaneous differentiation. Cell Prolif 42(1): 63–74. doi: 10.1111/j.1365-2184.2008.00571.x
- Pattappa G, Thorpe SD, Jegard NC, Heywood HK, de Bruijn JD, and Lee DA (2013). Continuous and uninterrupted oxygen tension influences the colony formation and oxidative metabolism of human mesenchymal stem cells. Tissue Eng Part C Methods 19(1): 68–79. doi: 10.1089/ten.TEC.2011.0734
- Goda N, and Kanai M (2012). Hypoxia-inducible factors and their roles in energy metabolism. Int J Hematol 95(5): 457–463. doi: 10.1007/s12185-012-1069-y
- Mahato B, Home P, Rajendran G, Paul A, Saha B, Ganguly A, Ray S, Roy N, Swerdlow RH, and Paul S (2014). Regulation of mitochondrial function and cellular energy metabolism by protein kinase C-λ/ι: a novel mode of balancing pluripotency. Stem Cells 32(11): 2880–2892. doi: 10.1002/stem.1817
- Pereira SL, Rodrigues AS, Sousa MI, Correia M, Perestrelo T, and Ramalho-Santos J (2014). From gametogenesis and stem cells to can- cer: common metabolic themes. Hum Reprod Update 20(6): 924–943. doi: 10.1093/humupd/dmu034
- Haneline LS (2008). Redox Regulation of Stem and Progenitor Cells. Antioxidants & Redox Signaling 10(11): 1849–1852. doi: 10.1089/ars.2008.2141
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, and Kume S (2014). Methionine me- tabolism regulates maintenance and differentiation of human pluripo- tent stem cells. Cell Metab 19(5): 780–794. doi: 10.1016/j.cmet.2014.03.017
- Wang J, Alexander P, Wu L, Hammer R, Cleaver O, and McKnight SL (2009). Dependence of mouse embryonic stem cells on threonine catabolism. Science 325(5939): 435–439. doi: 10.1126/science.1173288
- Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, Xu Z, Fischer KA, Devi A, Detraux D, Gu H, Battle SL, Showalter M, Valensisi C, Bielas JH, Ericson NG, Margaretha L, Robitaille AM, Margineantu D, Fiehn O, Hockenbery D, Blau CA, Raftery D, Margolin AA, Hawkins RD, Moon RT, Ware CB, and Ruohola-Baker H (2015). The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat Cell Biol 17(12): 1523–1535. doi: 10.1038/ncb3264
- Vernardis SI, Terzoudis K, Panoskaltsis N, and Mantalaris A (2017). Human embryonic and induced pluripotent stem cells maintain phe- notype but alter their metabolism after exposure to ROCK inhibitor. Sci Rep 7(1): 42138. doi: 10.1038/srep42138
- Prigione A, Fauler B, Lurz R, Lehrach H, and Adjaye J (2010). The senescence-related mitochondrial/oxidative stress pathway is re- pressed in human induced pluripotent stem cells. Stem Cells 28(4): 721–733. doi: 10.1002/stem.404
- Cho YM, Kwon S, Pak YK, Seol HW, Choi YM, Park DJ, Park KS, and Lee HK (2006). Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem Biophys Res Commun 348(4): 1472– 1478. doi: 10.1016/j.bbrc.2006.08.020
- Folmes CDL, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, and Terzic A (2011). Somatic oxida- tive bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 14(2): 264–271. doi: 10.1016/j.cmet.2011.06.011
- Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, Herrerías A, Batchelder EM, Plongthongkum N, Lutz M, Berggren WT, Zhang K, Evans RM, Siuzdak G, and Izpisua Belmonte JC (2012). The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res 22(1): 168– 177. doi: 10.1038/cr.2011.177
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, Asara JM, Daley GQ, and Cantley LC (2013). Influence of threonine metabo- lism on S-adenosylmethionine and histone methylation. Science 339(6116): 222–226. doi: 10.1126/science.1226603
- Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, Bomze D, Elena- Herrmann B, Scherf T, Nissim-Rafinia M, Kempa S, Itskovitz-Eldor J, Meshorer E, Aberdam D, and Nahmias Y (2015). Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differ- entiation of embryonic stem cells. Cell Metab 21(3): 392–402. doi: 10.1016/j.cmet.2015.02.002
- Vazquez-Martin A, Cufi S, Corominas-Faja B, Oliveras-Ferraros C, Vellon L, and Menendez JA (2012). Mitochondrial fusion by pharmaco- logical manipulation impedes somatic cell reprogramming to pluripo- tency: new insight into the role of mitophagy in cell stemness. Aging 4(6): 393–401. doi: 10.18632/aging.100465
- Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, and Melton DA (2008). Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Bio- technol 26(7): 795–797. doi: 10.1038/nbt1418
- Shyh-Chang N, and Daley GQ (2015). Metabolic switches linked to pluripotency and embryonic stem cell differentiation. Cell Metab 21(3): 349–350. doi: 10.1016/j.cmet.2015.02.011
- Johnson MT, Mahmood S, and Patel MS (2003). Intermediary metabolism and energetics during murine early embryogenesis. J Biol Chem 278(34): 31457–31460. doi: 10.1074/jbc.R300002200
- Mandal S, Lindgren AG, Srivastava AS, Clark AT, and Banerjee U (2011). Mitochondrial function controls proliferation and early differ- entiation potential of embryonic stem cells. Stem Cells 29(3): 486–495. doi: 10.1002/stem.590
- Pereira SL, Grãos M, Rodrigues AS, Anjo SI, Carvalho RA, Oliveira PJ, Arenas E, and Ramalho-Santos J (2013). Inhibition of mitochondrial complex III blocks neuronal differentiation and maintains embryonic stem cell pluripotency. PLoS ONE 8(12): e82095. doi: 10.1371/journal.pone.0082095
- Yang Y et al. (2017). Derivation of Pluripotent Stem Cells with In Vivo Embryonic and Extraembryonic Potency. Cell 169(2): 243-257.e25. doi: 10.1016/j.cell.2017.02.005
- Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, and Knoblich JA (2013). Cerebral organoids model human brain development and microcephaly. Nature 501(7467): 373–379. doi: 10.1038/nature12517
- Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch- Bräuninger M, Lewitus E, Sykes A, Hevers W, Lancaster M, Knoblich JA, Lachmann R, Pääbo S, Huttner WB, and Treutlein B (2015). Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci USA 112(51): 15672– 15677. doi: 10.1073/pnas.1520760112
- Eiraku M, and Sasai Y (2012). Self-formation of layered neural structures in three-dimensional culture of ES cells. Curr Opin Neuro- biol 22(5): 768–777. doi: 10.1016/j.conb.2012.02.005
- Amin ND, and Paşca SP (2018). Building Models of Brain Disor- ders with Three-Dimensional Organoids. Neuron 100(2): 389–405. doi: 10.1016/j.neuron.2018.10.007
ACKNOWLEDGMENTS
We sincerely apologize to colleagues whose work has not been included in this review due to space limitations. We would like to thank Natalya Orto-lano for edits and comments to this manuscript and all members of the Gama laboratory for their experimental and conceptual contributions, which led to some of the ideas presented in this review. We especially thank Me-gan Rasmussen for the design of all the illustrations used in this review.
This work was supported by NIH Grants: 5R00CA178190-05 (to VG), 1R35GM128915-01 (to VG), 1R21CA227483-01A1 (to VG) and 5T32ES007028-44 (to PJ).
COPYRIGHT
© 2019

Remodeling of mitochondrial morphology and function: an emerging hallmark of cellular reprogramming by Rastogi et al. is licensed under a Creative Commons Attribution 4.0 International License.


