Research Articles:
Cell Stress, Vol. 8, No. 1, pp. 125 - 139; doi: 10.15698/cst2024.12.302
Cold exposure reinstates NAD+ levels and attenuates hepatocellular carcinoma
1 Growth Factors, Nutrients and Cancer Group, Molecular Oncology Programme, Centro Nacional de Investigaciones Oncológicas (CNIO), Madrid, ES28029, Spain. 2 Instituto de Investigación Sanitaria HM Hospitales (IISHM), Madrid, Spain. 3 Laboratory of Innovation in Oncology, Gynecological, Genitourinary and Skin Cancer Unit, HM CIOCC, Centro Integral Oncológico Clara Campal, Hospital Universitario HM Sanchinarro, HM Hospitales, Madrid, ES-28050, Spain. 4 Department of Pathology, Hospital Universitario Ramón y Cajal, IRYCIS, Madrid, ES28034, Spain. 5 Universidad de Alcalá, Madrid, ES28801, Spain. 6 Histopathology Core Unit, Biotechnology Programme, CNIO, Madrid, ES-28029, Spain.
Keywords: cold exposure, hypthermia, NASH, HCC, NAD+, cell cycle, G2/M.
Abbreviations:
ALT - alanine aminotransferase,
AMPK - AMP-activated protein kinase,
BAT - brown adipose tissue,
CBT - core body temperature,
CT - cold temperature,
HCC - hepatocellular carcinoma,
NASH - non-alcoholic steatohepatitis,
RT - room temperature,
URI - unconventional prefoldin RPB5 interactor,
WAT - white adipose tissue.
Received originally: 28/11/2023 Received in revised form: 03/12/2024
Accepted: 05/12/2024
Published: 19/12/2024
Correspondence:
Nabil Djouder, Tel.: 0034 91 7328000 (Ext.: 3830); ndjouder@cnio.es
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Tatiana P Grazioso, Maria del Mar Rigual, Cristian Perna, Eduardo J Caleiras, Nabil Djouder (2024). Cold exposure reinstates NAD+ levels and attenuates hepatocellular carcinoma. Cell Stress 8: 125-139. doi: 10.15698/cst2024.12.302
Abstract
Cold exposure has been historically used for medicinal purposes, but its benefits and associated mechanisms in mammalian organisms still remain unclear. Here, we explore the chemoprotective properties of cold temperature using a mouse model of hepatocellular carcinoma (HCC) that recapitulates several human features. Chronic cold exposure is shown to prolong lifespan in diseased mice, enhance liver health, and suppress the development of aggressive HCC, preventing hepatocellular hypertrophy, high-grade oval cell hyperplasia, liver steatosis, and aberrant hepatocyte hyperproliferation. Mechanistically, exposure to cold temperatures reinstates NAD+ levels in the HCC mouse models that originally exhibited low NAD+ levels, a contributing process to the development of liver tumors. These findings uncover the role of cold therapy to attenuate HCC development and potentially other existing malignancies involving NAD+ modulation.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common type of primary malignant tumor that arises from hepatocytes, the main functional cells of the liver 1. It is a leading cause of cancer-related deaths worldwide, with limited treatment options available 2. While various therapeutic strategies have been explored, the development of effective treatments for HCC remains challenging. The use of cold exposure as a therapeutic intervention has shown promising results in various medical conditions, including cancer 3. However, the potential therapeutic benefits of cold exposure in HCC have not been thoroughly investigated.
The unconventional prefoldin RPB5 interactor (URI) has been demonstrated to function as an oncogene in mouse hepatocytes when overexpressed, URI inhibits aryl hydrocarbon (AhR)-and estrogen receptor (ER)-mediated transcription of enzymes implicated in L-tryptophan/kynurenine catabolism pathway leading to the de novo synthesis of nicotinamide adenine dinucleotide (NAD+). The deficits of NAD+ mediated by URI overexpression leads to DNA damage, replicative stress, inflammation, liver injury, and fibrosis, ultimately triggering non-alcoholic steatohepatitis (NASH) and aggressive HCC 4, 5, 6. Accordingly, nicotinamide riboside, a vitamin B3 derivative and booster of NAD+, prevents NASH-induced HCC 5, 6, 7. Interestingly, NAD+ plays a vital role in thermogenesis, a well-known process by which the body generates heat required for thermoregulation. Cold exposure can also trigger the activation of white adipose tissue (WAT) to increase heat production and preserve thermoneutrality 8, 9. Yet, the link between cold exposure, NAD+, and HCC development has not been elucidated so far.
In this study, we aim to explore the therapeutic potential of cold exposure on NAD+ levels and HCC development. We investigate the effects of cold on tumor growth, aggressiveness, and the underlying molecular mechanisms involved. Additionally, we evaluate the impact of cold exposure on key hallmarks of HCC, such as inflammation, cellular metabolism, and tumor progression. Understanding the therapeutic benefits of cold exposure in HCC could provide new insights into the development of novel treatment approaches. It may offer a non-invasive and potentially effective strategy for managing HCC, particularly for patients who are not suitable candidates for existing treatment modalities.
RESULTS
Hepatocytic hURI overexpression sensitizes mice to cold-induced stress
To study HCC in mice, we used a genetically engineered Col1a1 knock-in mouse model previously generated in our laboratory 5. This model enables the specific overexpression of human URI (hURI) in hepatocytes through the tetracycline-dependent transactivator (tTA) system, which expression is driven by the liver-activated protein promoter (LAP), yielding hURI-tetOFFHep mice, hereafter referred to as hURIHep (Figure 1A) 5. Absence of doxycycline results in the overexpression of hURI since conception. Overexpression of hURI was confirmed by immunohistochemistry (IHC) in hURIHep mouse liver tissues (Figure 1B).
–
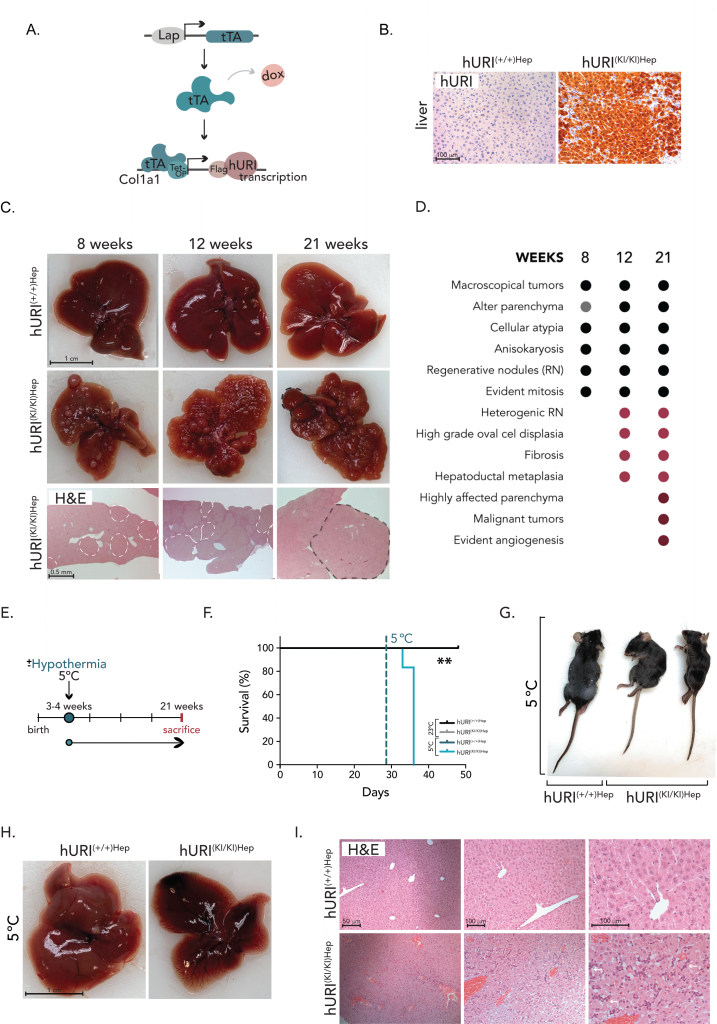 |
FIGURE 1: Hepatocytic hURI overexpression sensitizes mice to cold-induced stress. (A) Schematic representation of hURI-tetOFFHep (hURIHep) mouse model in which hURI expression is under the control of the hepatocyte-specific LAP promoter. (B) Representative pictures of immunohistochemistry (IHC) staining from liver sections of hURIHep mice highlighting hURI overexpression. (C) Representative images of livers from 8-,12-, and 21-week-old mice and representative pictures of hematoxylin and eosin (H&E) staining’s from hURI(+/+)Hep and hURI(KI/KI)Hep mice. (D) Histopathological characterization of 8-,12-, and 21-week-old hURI(KI/KI)Hepmice. (E) Scheme of hURIHep mice exposed to room temperature (RT) (23±1◦C) or cold temperature (CT) (5±1◦C) after weaning. (F) Survival curve of hURI(+/+)Hep [n=4], hURI(KI/KI)Hep [n=3] mice exposed to RT, and hURI(+/+)Hep [n=3] and hURI(KI/KI)Hep [n=6] mice exposed to CT. (G) Representative image of hURIHep mice exposed to RT and CT. (H) Representative images from hURIHep mice livers exposed to CT. (I) Representative pictures of H&E from liver sections of hURIHep mice exposed to CT. White arrows highlight oval cell hyperplasia. Statistical analysis was performed using the Kaplan-Meier curve (F). Unless otherwise indicated, data are represented as mean ± s.e.m from “n” independent experiments; **P ≤0.01. Scale bars, 50 µm, 100 µm (B, I), 0.5 mm (C), and 1 cm (C, H). |
Heterozygous hURI overexpression (hURI(+/KI)) in mouse hepatocytes reportedly results in a two-fold increase compared to endogenous mouse URI (mURI) and promotes the spontaneous development of macroscopic lesions including high grade dysplastic nodules and adenomas by 32 weeks of age 5. In contrast, homozygous hURI expression (hURI(KI/KI)Hep mouse), which is six-fold higher than endogenous mURI, accelerates tumor development, starting at 8 weeks of age (Figure 1C-D). Initially, at 4 weeks of age, hURI(KI/KI)Hep mice exhibited no macroscopical apparent signs of pathology or tissue alterations in the liver. At 8 weeks, macroscopic tumors were apparent, and livers showed an abnormal parenchyma characterized by cellular atypia, anisokaryosis, and few regenerative nodules with evident mitosis. By 12 weeks, hURI(KI/KI)Hep mice displayed anisokaryosis, multiple heterogeneous regenerative nodules and small adenomas, high-grade oval cell proliferation, hepatoductal metaplasia, and fibrosis, all standard features occurring at early stages of hepatocarcinogenesis. Lastly, at 21 weeks, hURI(KI/KI)Hep mice presented a highly affected parenchyma with fully developed HCC and noticeable angiogenesis (Figures 1C-D). Conversely, control hURI(+/+)Hep mice displayed a normal liver architecture with no signs of pathology throughout the 4–21-week period (Figures 1C).
To study the properties of cold temperature (CT), we used the Memmert climate chamber, which allows mice to be housed at various temperatures ranging from 5°C to 40°C. First, we designed a pilot study in which C57BL/6 mice were housed for one month at either normal room temperature (RT) (23 ±1°C) or CT (5±1°C) to evaluate their ability of mice to cope with long-term cold exposure and to test the efficiency of our equipment. Mice were monitored daily, and no signs of pain, suffering, or distress were observed. Additionally, no clinical symptoms, weight loss, or mortality were detected, indicating that C57BL/6 mice can tolerate significant temperature changes (data not shown).
To elucidate the role of temperature in tumorigenesis, we conducted long-term cold exposure treatments on hURIHep mice using homozygous hURI overexpression to accelerate tumorigenesis (Figure 1E). Weaned 3-week-old hURIHep mice were housed for one month either at RT (23±1°C) or at CT (5±1°C), to assess the ability of hURIHep mice to tolerate prolonged exposure to cold (Figure 1E). Throughout the experiment, hURIHep mice at CT or RT were monitored daily. Strikingly, hURI(KI/KI)Hep mice exposed to CT experienced a high mortality rate of 100%, with death occurring within 8 to 13 days after being housed at CT, while hURI(+/+)Hep mice exposed to either CT or RT and hURI(KI/KI)Hep mice at RT survived (Figure 1F) with no indications of pain, suffering, or distress (Figure 1G).
Based on these observations and to further understand why hURI(KI/KI)Hep mice were not able to cope with cold, we exposed weaned hURIHep mice to 5°C and sacrificed them six days after cold exposure (Sup Figure 1A). Through the length of the treatment, one hURI(KI/KI)Hep mouse died five days after cold exposure, supporting the lethality of CT upon hURIHep overexpression (Sup Figure 1B). In addition, we monitored weight and observed that all hURI(KI/KI)Hep mice lost a significant percentage of body weight upon cold when compared to their control hURI(+/+)Hep littermates (Sup Figure 1C). Furthermore, histological characterization revealed a high grade of liver damage in hURI(KI/KI)Hep mice (Sup Figure 1D). In concordance, we assessed hepatic function and liver injury by measuring specific blood liver markers and observed that hURI(KI/KI)Hep mice presented high levels of alkaline phosphatase (ALP) and alanine aminotransferase (ALT); and decreased bile acid levels (Sup Figure 1E), when compared to its control hURI(KI/KI)Hep littermates exposed to cold, supporting that hURI(KI/KI)Hep mouse livers were functionally damaged. These results may indicate that hURI(KI/KI)Hep mice died from fulminant liver failure upon cold-shock exposure, while hURI(+/+)Hep mice were completely healthy when exposed to low temperatures. However, these are very preliminary data, where the experimental set number must be increased, and further metabolic and biological studies are required to fully understand why overexpression of hURIHep in mouse hepatocytes leads to early death upon cold.
Next, histopathological post-mortem examinations of hURI(KI/KI)Hep mouse livers exposed to CT for two weeks (Figure 1E) revealed disrupted tissue architecture with significant hepatic and lobule damage compared with hURI(+/+)Hep mouse livers (Figures 1G-I). Additionally, hURI(KI/KI)Hep mouse livers exhibited localized hepatocyte necrosis and apoptosis, marked by the presence of apoptotic bodies, as well as oval cell hyperplasia (Figure 1H-I). Pathological assessment indicated that livers of dead mice had a high degree of lymphocyte infiltration with inflammatory foci and an excessive accumulation of iron deposits (Figure 1H-I), suggesting impaired liver function. Moreover, areas of autolysis and extensive destruction of red blood cells were detected, marking signs of severe liver injury (Figure 1H-I). Notably, cold exposure was lethal in hURI(KI/KI)Hep mice, highlighting a possible vital role of URI in body homeostasis regulation and response to cold-induced stress.
However, it is important to consider that high levels of DNA damage and low hepatic NAD+ levels were already present in hURI(KI/KI)Hep mutant mice 5 when exposed to cold. This suggests that the body’s NAD+ reserves were preferentially utilized for thermoregulation rather than DNA repair, reflecting energy trade-offs. Consequently, the combination of impaired physiological homeostasis, liver failure, and severe DNA damage due to URI overexpression may be detrimental and lethal for hURI(KI/KI)Hep
mice.
Lifespan-enhancing properties of cold in hURI overexpressing mice
To overcome this problematic, and as most mammals in nature undergo adaptive and behavioral adjustments to cope with temperature variations 10, thereby enhancing their resistance and tolerance to temperature changes, we suggested that an acclimatization period would help animals achieve physiological stability and restore homeostasis before CT. Thus, prior to chronic CT exposure, we subjected weaned hURI(+/+)Hep and hURI(KI/KI)Hep mice to an acclimatization period, during which mice were housed at 18°C for one week. Following this acclimatation phase, the ambient temperature was gradually decreased to 5°C, and mice were continuously exposed to CT until they were sacrificed at 21 weeks of age (Figure 2A).
In parallel, control mice (both hURI(+/+)Hep and hURI(KI/KI)Hep) were housed at RT and sacrificed at 21 weeks of age (Figure 2A). All hURI(+/+)Hep mice, regardless of whether they were housed at RT or CT, survived the entire duration of the experiment (Figure 2B). Notably, the acclimatization period enabled hURI(KI/KI)Hep mice to adapt and cope with CT, thereby preventing early death. Remarkably, hURI(KI/KI)Hep mice exhibited an improved survival rate when exposed to CT when compared to those housed at RT (Figure 2B). Approximately 25% of hURI(KI/KI)Hep mice at RT died within four and six weeks, while exposure to CT decreased the mortality rate to 16% and extended the life expectancy by approximately six weeks (Figure 2B). Thus, exposure to cold temperatures may have a beneficial effect on lifespan extension in diseased hURI(KI/KI)Hep mice. This observation aligns with previous studies that correlate low temperatures with enhanced lifespan across a range of organisms, from poikilothermic to homeothermic species 3, 9, 11.
–
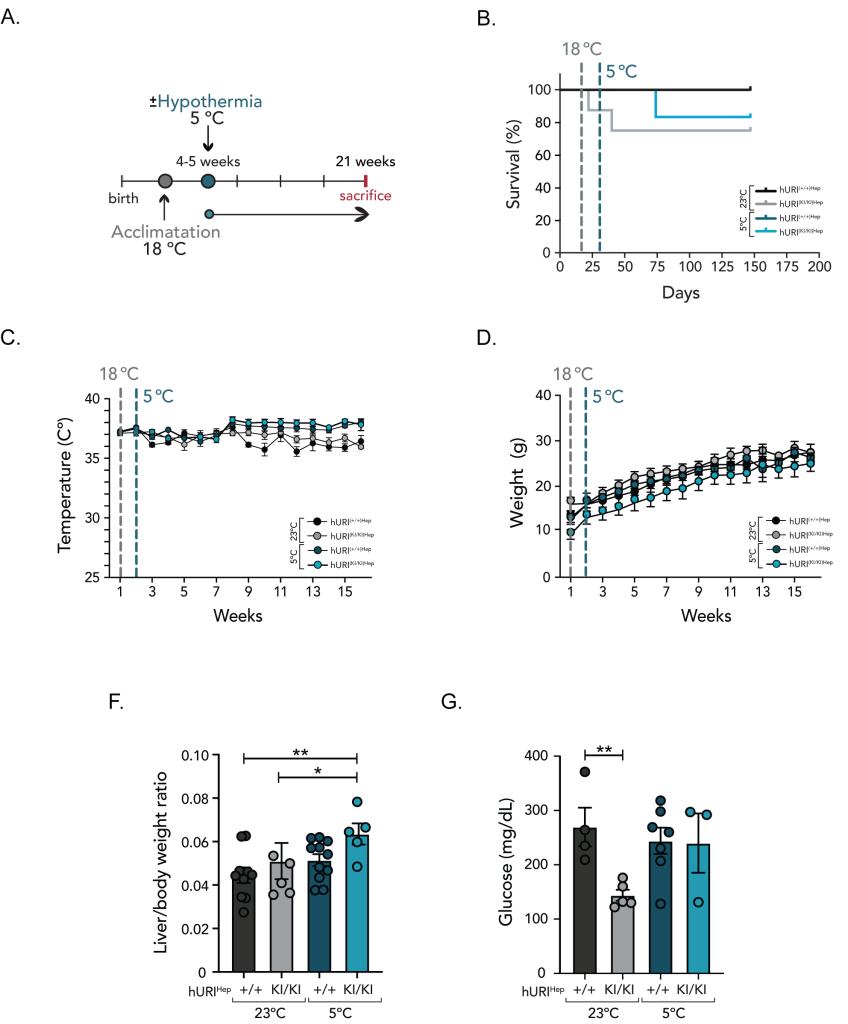 |
FIGURE 2: Lifespan-enhancing properties of cold in hURI overexpressing mice. (A) Scheme of hURIHep mice exposed to CT. Mice were exposed to an acclimatization period (one week at 18◦C after weaning) followed by CT (18 weeks at 5±1◦C). Control hURIHep mice were exposed to RT (23±1◦C). hURIHep mice were sacrificed at 21 weeks of age. (B) Survival curve of hURI(+/+)Hep [n=11], hURI(KI/KI)Hep [n=8] mice exposed to normal RT conditions, and hURI(+/+)Hep [n=11] and hURI(KI/KI)Hep [n=6] mice exposed to CT. (C) Core body temperature record of hURI(+/+)Hep [n=11], hURI(KI/KI)Hep [n=8] mice exposed to normal RT conditions, and hURI(+/+)Hep [n=11] and hURI(KI/KI)Hep [n=6] mice exposed to CT. (D) Weekly measurements of hURI(+/+)Hep [n=11], hURI(KI/KI)Hep [n=8] mice exposed to normal RT conditions and hURI(+/+)Hep [n=11] and hURI(KI/KI)Hep [n=6] mice exposed to CT. (E) Liver to body weight ratio of hURI(+/+)Hep [n=10], hURI(KI/KI)Hep [n=6] mice exposed to normal RT conditions, and hURI(+/+)Hep [n=11] and hURI(KI/KI)Hep [n=5] mice exposed to CT. (F) Glucose in blood of hURI(+/+)Hep [n=4], hURI(KI/KI)Hep [n=5] mice exposed to normal RT conditions, and hURI(+/+)Hep [n=7] and hURI(KI/KI)Hep [n=3] mice exposed to CT. Statistical analyses were performed using Kaplan-Meier curve (B) or multiple comparison ANOVA test (E-F). Unless otherwise indicated, data are represented as mean ± s.e.m from n independent experiments; *P ≤ 0.05, **P ≤ 0.01. |
Furthermore, core body temperature (CBT) was monitored weekly by using a rectal probe thermometer. Interestingly, after six weeks of CT, both hURI(+/+)Hep and hURI(KI/KI)Hep mice exhibited higher CBT when compared to their littermates housed at RT (Figure 2C), which may imply that URI activity is independent of thermoregulation since both hURI(+/+)Hep and hURI(KI/KI)Hep mice exposed to CT enhanced their thermogenic capacity. Weekly weight monitoring revealed no significant differences in body weight among hURI(+/+)Hep mice when exposed to either RT or CT, but weight differences were observed between hURI(KI/KI)Hep mice housed at RT and hURI(KI/KI)Hep mice kept at CT (Figure 2D).
Full-body necropsies showed that when hURI(KI/KI)Hep mice were subjected to CT and sacrificed at 21 weeks of age, they exhibited a higher liver-to-body weight ratio when compared to hURI(KI/KI)Hep mice maintained at RT (Figure 2F). However, no changes in brown adipose tissue (BAT) tissue weight were observed upon CT exposure, and no weight differences nor apparent anatomical abnormalities were observed in the rest of the organs (data not shown). Notably, hURI(KI/KI)Hep mice exposed to CT also restored blood glucose levels when compared to their counterparts at RT (Figure 2G).
Long-term cold exposure attenuates HCC development
Macroscopic tissue inspection and histopathological evaluation of mouse livers revealed no apparent signs of liver pathology in hURI(+/+)Hep mice. Interestingly, all hURI(KI/KI)Hep mice developed tumors, mainly localized in the visceral surface of the liver, however, hURI(KI/KI)Hep mice exposed to CT significantly abolished the development of aggressive HCC when compared to their control hURI(KI/KI)Hep mice exposed to RT which developed up to grade 4 adenocarcinomas (Figure 3A). Moreover, CT significantly reduced the proportion of damaged liver tissue area (damage/healthy tissue), and tumor incidence and tumor burden were significantly decreased upon CT in hURI(KI/KI)Hep mice when compared to its control hURI(KI/KI)Hep littermates at RT (Figures 3A-E), hinting, cold-protective properties that ameliorate tumor growth and aggressive HCC development.
–
FIGURE 3: Long-term cold exposure attenuates HCC development. (A) Representative images of livers from mice exposed to RT (23±1◦C) or CT (5±1◦C). White dashed lines highlight regenerative nodules. (B) Percentage of hURIHep mice bearing liver tumors. (C-E) Macroscopical characterization of liver tumors, representing (C) percentage of affected area, (D) number of tumors, and (E) tumor size in hURIHep mouse livers exposed to RT (23±1◦C) or CT (5±1◦C). (F) Representative H&E stainings from liversof hURIHep mice exposed to RT or CT. (G-H) Microscopical quantification of the percentage of hURIHep mice presenting (G) hepatocytehypertrophy and (H) oval cell hyperplasia, based on liver tissues from (F). (I) Microscopical quantification of the percentage of hURIHep mice presenting steatosis based on liver tissues from (F). (J) Representative H&E staining’s from hURI(KI/KI)Hep mice livers. White arrows highlight oval cell hyperplasia and black arrows highlight anisokaryosis. White dashed lines highlight cholestasis, while the white circle shows a nodule with clear cells. (K) Representative H&E staining’s from hURI(KI/KI)Hepmice livers highlighting steatosis. White dashed lines highlight hepatocytes with microvesicular steatosis and black arrows show macrovesicular steatosis. (L) Western blot analysis of hURIHep mouse livers exposed to RT or CT. Statistical analysis was performed using unpaired two-tailed Student’s t-test. Data are represented as mean ± s.e.m from n independent experiments; *P≤ 0.05, **P ≤ 0.01. Scale bars, 50 µm and 100 µm(F, J,K), and 1 cm (A). |
Further histopathological evaluation of mouse livers by H&E showed that the severity of architectural abnormalities was more pronounced in hURI(KI/KI)Hep mice kept at RT, as evidenced by the presence of lobular and portal irregularities along with central vein dysplasia, sinusoidal compression, and increased hepatocellular hypertrophy, which was ameliorated in mice exposed to CT (Figures 3F-3G, 3J). Furthermore, high-grade oval cell hyperplasia was ameliorated upon CT, and while approximately 80% of hURI(KI/KI)Hep mouse livers at RT presented microvesicular steatosis (small lipid vesicles in the cell cytoplasm) and macrovesicular steatosis (one large vacuole of lipids within the cell cytoplasm) – a condition frequently linked to various liver pathologies triggering lipotoxicity, inflammation, cell death and metabolic dysfunction – it was observed that CT was sufficient to completely revoked steatosis (Figures 3F, 3H, 3J-K). Although tumors were present in all hURI(KI/KI)Hep mice, those treated with cold were protected against the development of aggressive HCC after 21 weeks. In contrast, hURI(KI/KI)Hep mice kept at RT did not experience such protection, suggesting that cold exposure inhibited malignant cell transformation and tumor growth, consistent with previous findings.
Furthermore, we checked common liver markers in hURI blood samples, to evaluate the metabolic function of the liver. hURI(KI/KI)Hep mice exposed to CT presented a decrease, though not significant, of alanine aminotransferase (ALT), gamma-glutamyltransferase (GGT), and bile acids levels when compared to hURI(KI/KI)Hep littermates at RT (data not shown), suggesting that cold may protect from liver injury preventing hepatic function impairment. Lastly, to validate the absence of malignant HCC tumors, we examined the expression of HSP70, a gene known to be highly upregulated in the early stages of HCC when compared to pre-neoplastic lesions, and commonly used as a molecular marker for differentiating HCC stages 12. The expression of HSP70 in the livers of hURI(KI/KI)Hep was significantly decreased in those mice exposed to CT when compared to hURI(KI/KI)Hep livers at RT (Figure 3L). These decrease in HSP70 expression is consistent with the absence of aggressive HCC tumors in hURI(KI/KI)Hep mice exposed to CT, providing evidence for cold-protective properties that contribute to the decreased development of aggressive liver tumors.
Cold-induced acetyl-CoA carboxylase inhibition
The initial event triggering malignant cellular transformation upon hURI overexpression in hepatocytes is the impairment of the L-tryptophan/kynurenine pathway. This impairment results in reduced NAD+ concentrations, leading to the induction of DNA damage, and subsequently HCC and NASH development 4, 5, 6, 12. The supplementation of nicotinamide riboside effectively boosts NAD+ pools, mitigating DNA damage, and thereby preventing HCC 4, 5, 6, 12. Moreover, NAD+ reduction has been associated with various age-related pathologies and restoring NAD+ levels has demonstrated the ability to ameliorate or prevent the onset of diverse diseases 13.
Considering that both CT and boosting NAD+ levels have positive therapeutic effects, we hypothesize that the link between the observed benefits of cold in HCC prevention might be NAD+. Therefore, we assessed hepatic NAD+ levels in 21-week-old mice exposed to RT and CT. Intriguingly, and in line with previous studies 5, NAD+ concentrations were significantly reduced in hURI(KI/KI)Hep mice exposed to RT. However, cold appears to reduce NAD+ concentrations in both hURI(+/+)Hep and hURI(KI/KI)Hep mouse livers (Figure 4A). This suggests a dynamic rather than static role of NAD+ that must be continuously synthesized, consumed, and/or recycled upon cold exposure to induce thermoregulation. To further examine whether NAD+ levels are modulated upon CT, we exposed HUH7, HepG2 and U2OS cell lines to 27°C (representing CT) and 37°C (representing RT) for 24 hours (Figure 4B).
–
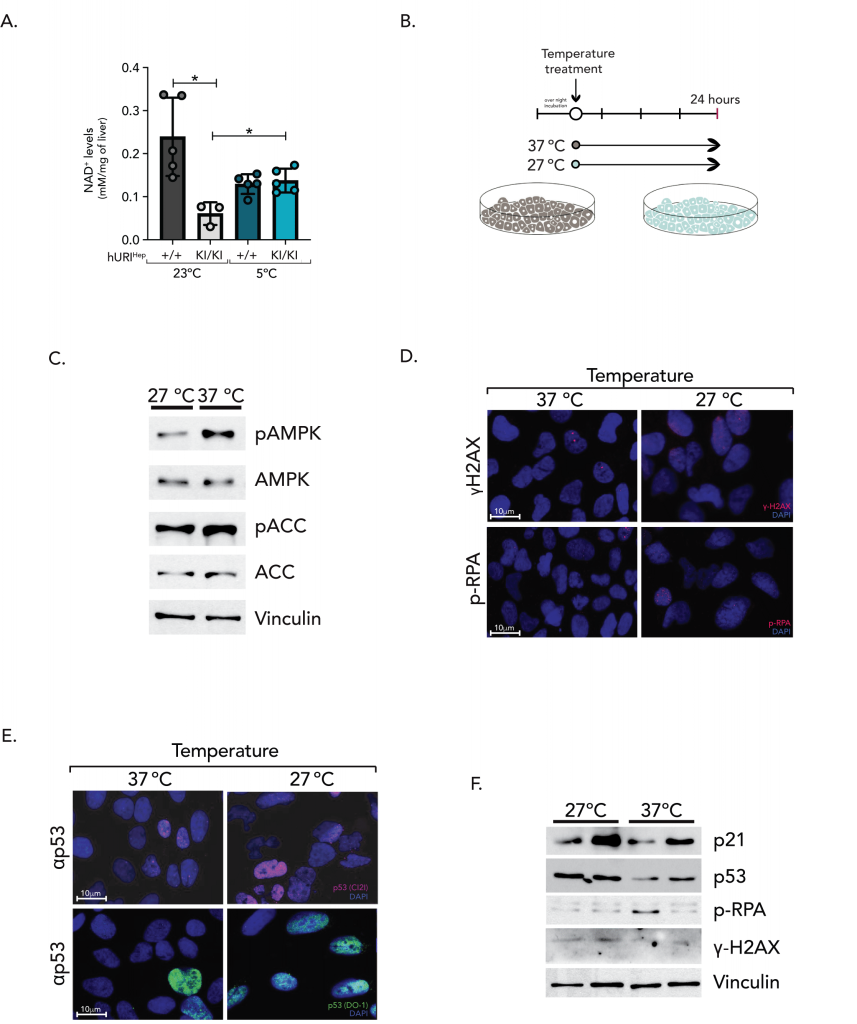 |
FIGURE 4: Cold-induced acetyl-CoA carboxylase inhibition. (A) NAD+ levels in 21-week-old hURIHep mouse livers exposed to RT or CT. (B) Scheme of U2O2 cells treated undernormal temperature (37◦C) or cold temperature (27◦C). (C) Western blot analysis of U2OS cells exposed for 24 hours to 27◦C or 37◦C. (D-E) Representative pictures of IF staining’s in U2OS cells incubated at 27◦C or 37◦C for 24 hours. (F) Western blot analysis of U2OS cells incubated at 27◦C or 37◦C for 24 hours. |
Given that NAD+ and NADH play a vital role in energy metabolism and homeostasis by supporting redox reactions, glycolysis, oxidative phosphorylation, and the mitochondrial electron transport chain (ETC) 14, 15, we evaluated the cellular energy status upon cold exposure. The AMP-activated protein kinase (AMPK) acts as an energy sensor, adjusting its activity according to the cell’s AMP:ATP ratio 16. Consequently, AMPK phosphorylation on Thr-172 (p-AMPK), which reflects cellular energy deficits, triggers catabolic processes while suppressing anabolic pathways. Moreover, AMPK has been shown to enhance SIRT1 activity by increasing NAD+ levels 17. Upon cold exposure, p-AMPK expression was significantly diminished (Figure 4C), when compared to cells maintained at 37°C, with a 0.39-fold decrease. Additionally, p-AMPK showed a 0.41-fold decrease relative to total AMPK expression, indicating intracellular energetic homeostasis, with high ATP levels and prevalent anabolic pathways. Upon cellular energy deficits, AMPK inhibits fatty acid synthesis and promotes fatty acid oxidation by phosphorylation of acetyl-CoA carboxylase (ACC 1/2) 16. However, no major differences were observed in its downstream effector ACC between cells exposed to 27°C and 37°C (27°C=0.85-fold change) (Figure 4C). Cold exposure led to a 2.12-fold increase in p-ACC, suggesting that ACC phosphorylation triggered by low temperatures is independent of AMPK activity, while ACC phosphorylation at 37°C is dependent on AMPK. This indicates that cold exposure may inhibit ACC-mediated fatty acid synthesis and trigger fatty acid oxidation and lipolysis to sustain thermoregulation. In agreement with this idea, the abolishment of steatosis observed in hURI(KI/KI)Hep mouse livers exposed to cold temperatures (Figures 3F, 3I, 3K) might be explained by increased fatty acid oxidation resulting from cold-induced ACC inhibition.
Moreover, since NAD+ levels were diminished upon cold, we wonder whether NAD+ was consumed to sustain thermoregulation and restore DNA integrity, explaining the less aggressive phenotype observed in hURI(KI/KI)Hep mouse livers. However, no discernible differences in DNA damage levels, as indicated by γH2AX-positive nuclei and phosphorylated RPA, were observed between cells under both conditions (Figure 4D-F). Nonetheless, there was an increase in p53 levels and its target gene p21 in the cells cultured at CT (Figure 4E-F). This increase may be attributed to the upregulation of the NAD+– SIRT1 axis, leading to p53 acetylation and stabilization. Considering that p53 and p21 are induced upon cold exposure independently of DNA damage, p53 and p21 might be involved in cell cycle arrest, senescence, or apoptosis.
Cold exposure induces p53/p21-dependent G2/M cell cycle arrest
As we observed that cold triggered p53 and p21 activity independently of DNA damage, we hypothesized that cold might impair the cell cycle and, consequently, proliferation. Thus, we checked common markers of proliferation. Notably, there was an increase in KI67-positive nuclei in the livers of hURI(KI/KI)Hep mice exposed to CT when compared to hURI(KI/KI)Hep mice exposed at RT, while the expression of proliferating cell nuclear antigen (PCNA) was reduced in their hepatocytes (Figure 5A-C). Since KI67 and PCNA are markers with distinct expression patterns throughout the cell cycle, where KI67 is continuously expressed from G1 through mitosis, while PCNA activity is absent during the G2/M phase 18, our data suggest that cold may induce G2/M cell cycle arrest, potentially orchestrated by p53 and p21. Consistent with this, the number of U2OS, HepG2, and HUH7 cells significantly decreased upon cold exposure when compared to cells maintained at 37°C, which typically exhibited an average exponential growth (Figure 5D-E), suggesting that cell proliferation is compromised upon cold. To further prove this idea, we examined cell cycle profiling by propidium iodide (PI). Cell cycle analysis revealed that cold exposure significantly increased G2/M phase, suggesting CT arrests cells prior entering mitosis, accompanied by a notable decrease in the G0/G1 phase (Figure 5F), supporting that cold may inhibit proliferation by impairing the cell cycle. In line with the observed decrease of PCNA and G2/M arrest, our results might suggest that hyperproliferation impairment may limit neoplastic lesions growth during HCC development in hURI(KI/KI)Hep mouse livers exposed to cold. Furthermore, given that cold induces G2/M cell cycle arrest, we investigated whether p53 promoted apoptosis in hepatocytes unable to complete the cell cycle. However, Western blot analysis did not detect the expression of the apoptosis marker cleaved caspase 3 (CC3) in the hepatocytes of hURIHep mice exposed to CT or RT (Figure 5C). Thus, our data suggest that cold compromises cell cycle progression, leading to impaired hyperproliferation and thus, tumor growth.
–
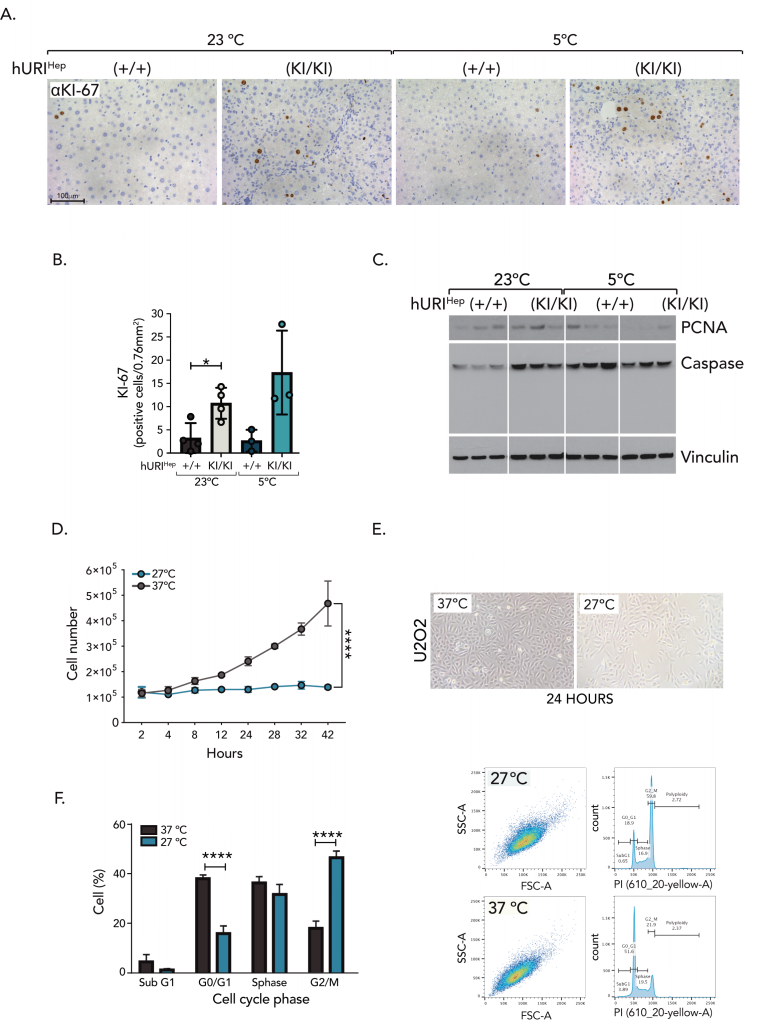 |
FIGURE 5: Cold exposure induces p53/p21-dependent G2/M cell cycle arrest. (A-B) (A) Representative pictures of KI67 IHC and (B) its quantification from liver sections of hURIHep mice exposed to RT or CT. (C) Western blot analysis of hURIHep mouse livers housed at RT or CT. (D) Cell number count of U2OS cells incubated for 24 hours at 27◦C or 37◦C [n=3]. (E) Representative image from bright field microscope of U2OS cells incubated for 24 hours at 27◦C or 37◦C. (F-G) Representative PI flow cytometry plots (F) and percentage of cell populations present at the different phases of the cell cycle (G). U2OS cells were incubated at 27◦C or 37◦C for 24 hours [n=3]. Statistical analysis was performed using Brown-Forsythe and Welch ANOVA (B), two-way ANOVA (D), and unpaired two-tailed Student’s t-test (F). Unless otherwise indicated, data are represented as mean ±s.e.m from n independent experiments; *P ≤ 0.05****P ≤ 0.001. Scale bars 100 µm (A). |
DISCUSSION
The therapeutic use of temperature has been practiced since ancient times 3, yet the underlying mechanisms responsible for its health-enhancing effects remain unclear. In our study, we investigated the protective properties of cold exposure (5°C) in an oncogene-induced HCC model. We first observed that drastic temperature changes can be lethal in mice overexpressing hURI. Given that high levels of DNA damage and low hepatic NAD+ levels are already present in hURI(KI/KI)Hep mutant mice, our data might suggest that NAD+ reservoirs in the body were utilized to cope with thermoregulation over DNA repair (energy tradeoffs). From a thermodynamic perspective, energy balance is defined by three components: (i) obligatory energy expenditure, necessary for the normal functioning of cells (basal metabolic rate); (ii) energy expenditure attributed to adaptive thermogenesis, required to maintain CBT (thermoregulation); and (iii) energy expenditure used during physical activity 19. Upon cold exposure, metabolic and physical activities are reduced, and behavioral mechanisms to conserve heat are activated. If these mechanisms are insufficient, energy expenditure rates required for heat production and thermoregulation increase to maintain thermal homeostasis 20, 21. Since NAD+ is essential for regulating whole-body energy metabolism and adaptive thermogenesis 9, 15, 22, we can speculate that DNA repair may participate in energetic trade-offs competing with thermoregulation for ATP and NAD+.
Consistent with the weight loss observed in hURI(KI/KI)Hep mutant mice, ATP, and NAD+ are likely utilized for thermoregulation rather than repairing the DNA damage present in the hepatocytes of young hURI(KI/KI)Hep mice. Consequently, persistent and irreparable DNA damage may induce genotoxic and oxidative stress, predisposing liver injury and dysfunction, thus favoring cold-induced mortality. Nevertheless, once hURIHep mutant mice were exposed to an acclimatization period, they were able to cope and survive cold exposure. Most likely, this period allowed mice to gradually adapt to drastic thermal changes leading to behavioral adjustments that reduce thermogenesis-induced metabolic cost during cold exposure 21, minimized distress, and favored cold tolerance 23.
Our results show how chronic cold exposure has health-enhancing effects, associated with an extended life expectancy of hURI(KI/KI)Hep mice, linked with a 9% decrease in mortality (RR: 0.66; mortality: RT=25%; CT=16.6%). In accordance with previous published data where cold exposure has previously been shown to enhance longevity 24, 25, 26, our study suggests that cold exposure may enhance life expectancy not only in healthy individuals but, also in cancer patients. Furthermore, we show that cold exposure exhibits antitumor activities by improving liver histopathological features which predispose tumorigenesis. Specifically, cold mitigated hepatocyte hypertrophy and oval cell expansion, which are known key contributors to the neoplastic transformation of hepatocytes 2. Interestingly, high levels of oval cell proliferation were present at earlier time points in 12-week-old hURI(KI/KI)Hep mice, which might lead us to believe that preventing hepatocytes malignant transformation upon cold exposure may be one of the first events protecting against tumor development. Lastly, we observe how cold exposure completely prevented the development of HCC in our model. Although all hURI(KI/KI)Hep mouse livers exhibited lesions, 100% of cold-treated mice remained HCC free, while hURIHep mutant mice at RT presented up to grade 4 adenocarcinomas with evident angiogenesis. However, it remains to be determined whether cold exposure entirely abolishes HCC development or only delays it, as tumor growth inhibition has been proved to be reversible upon the cessation of low-temperature exposure 8.
Mechanistically, we observed that CT significantly increases NAD+ levels in hURI(KI/KI)Hep mice, which originally had de novo NAD+ levels affected triggering HCC development 5. This suggests that the elevation of NAD+ levels upon CT could play a protective role against hepatocarcinogenesis. Interestingly, CT does not completely restore NAD+ levels to those seen in control mice at RT. However, considering that NAD+ is required more than 500 enzymatic reactions, it is involved in multiple chemical and biological processes 7, 27, 28; and since NAD+ concentrations are not statistic and depend on the cellular needs and the regulatory activity of NAD+-synthesizing and NAD+-consuming enzymes (such as PARPs, sirtuins and CD38) 29. This discrepancy can be attributed to other metabolic reactions and rewiring necessary for the activity of NAD+, in that matter low levels of NAD+ upon cold might reflect its exacerbated consumption. For instance, cold exposure has been shown to activate thermogenesis by triggering WAT lipolysis and the activation of the Uncoupling protein-1 (UCP1) 30, leading to NAD+ regeneration. Additionally, NAMPT and the BAT-specific apoptosis-inducing mitochondrion-associated factor 2 (Aifm2) have been shown to be induced upon cold exposure, fasting, and ß-adrenergic stimuli, promoting NAD+ synthesis 15, 22, and Aifm2 in fact, has been shown to regenerate NAD+ to support UCP1 thermogenesis in the mitochondria 15. This heightened metabolic activity assists in heat generation and the maintenance of CBT9. Taken together, this might suggest that NAD+ is constantly consumed (hence, explaining the low levels observed in hURI(KI/KI)Hep mice at CT), but at the same time, its synthesis is enhanced in response to cold exposure, thus, constantly, produced-consumed-recycled NAD+ may aid both in DNA repair, protecting from lived damage and thus, tumor development, and thermoregulation. Further work will certainly decipher how cold triggers Aifm2 activity in the liver, thereby enhancing local NAD+ synthesis.
Recent studies indicate that cold exposure and cold acclimation offer various beneficial effects beyond thermogenesis 3, 9. These effects include improved insulin sensitivity, increased energy expenditure, enhanced glucose and lipid metabolism, and anti-inflammatory effects. It is noteworthy that hURI(KI/KI)Hep mice are diabetic 5; however, their blood glucose levels were normalized upon cold exposure, suggesting that NAD+ may be regenerated to support robust glycolysis. Nevertheless, the significance of NAD+ in cold-induced protection cannot be dismissed, as it plays a crucial role in energy metabolism.
Our findings indicate that cold exposure prevents hepatic steatosis, potentially by promoting lipolysis and fatty acid oxidation through ACC inhibition. This supports previous studies showing that ACC1/2 depletion and pharmacological ACC inhibition can reduce hepatic steatosis, tumor formation, lipid accumulation, hepatic stellate cell activation, and fibrosis 31, 32. Consistently, cold exposure completely abolished hepatic steatosis in hURI(KI/KI)Hep mouse livers, likely through ACC inhibition. Thus, cold-induced ACC inhibition may support lipolysis and thermogenesis while offering tumor-protective properties.
Cold exposure induces G2/M cell cycle arrest, likely through the upregulation of the p53/p21 pathway, preventing hepatocyte hyperproliferation, a key factor in tumor development 12. Consistent with our findings, p53 activation by cold-shock treatments have been shown to repress cyclin B1 and cdc25 or transactivate p21, leading to G2/M arrest 33. This results in the inhibition of CDK1, CDK2, and PCNA, observed in hURI(KI/KI)Hep mouse livers. Our study did not observe apoptosis, suggesting p53/p21 activation may cause a transient checkpoint arrest for DNA repair or irreversible senescence. Thus, cold exposure could serve as a less aggressive approach to inhibit hyperproliferation and enhance conventional cancer therapies.
In conclusion, our study demonstrates that chronic cold temperature exposure exerts protective properties against HCC in mice. The observed health-enhancing effects include an extension of life expectancy. These findings open avenues for exploring cold exposure therapies as a less aggressive adjuvant therapeutic approach to ameliorate HCC and enhance the efficacy of conventional cancer therapies.
MATERIALS AND METHODS
Antibodies
Antibodies used for immunoblotting IHC and IF were as followed: Mouse monoclonal anti-Vinculin (#V9131) (WB:1:5000) and Phospo-Histone H2A.X (Ser139) (#05- 636) (WB: 1:2500, IHC/IF: 1:500) were purchased from Merck-Millipore. Mouse monoclonal p21 (#556430) (WB: 1:1000) was obtained from BD Biosciences. p53 (D0-1) (#18032) (WB: 1:1000, IF: 1:500), Cleaved caspase-3 (CC3) (Asp-175) (#9661) (WB: 1:100; IHC:1:250), AMPK (#2532) (WB: 1:1000), p-AMPK (#2535s) (WB: 1:500), ACC (#3662) (WB: 1:1000), p-ACC (ser-79) (#366IL) (WB: 1:1000) and p-RPA (Ser8) (#83845) (WB: 1:1000, IF: 1:500) were purchased from Cell signaling Technology. Rabbit polyclonal p53 (CM5) (#VP-P956) (WB: 1:1000, IF: 1:500) was obtained from Vector Laboratories. Rat monoclonal anti-PCNA (PC10) (#sc-56) (WB: 1:500) and mouse monoclonal anti-Hsp70 (clone W27) (#sc-24) (WB: 1:1000) were obtained from Santa Cruz Biotechnology. Rabbit polyclonal anti-KI67 (SP6) (#MAD-000310QD) (IHF/IF: non-diluted) antibody was purchased from Master Diagnostica. hURI (WB: 1:500, IHC: 1:200) was produced by Dr. Nabil Djouder as previously described 34, 35.
Mouse models
The hURI knock-in mouse model (hURI-tetOFFHep) was generated as previously described 5. Hepatocyte specific hURI overexpression was generated by crossing a Col1a1 hURI mouse with a line containing the tetracycline-dependent transactivator (tTA) under the control of the liver activated protein (LAP) promoter breeding LAP-tTA/hURItetOFF mouse, here referred to as hURIHep mice. Upon doxycycline administration, specific hURI expression can be switched off. However, in our study, hURI-tetOFFHep mice were not treated with doxycycline, so hURI was expressed since conception. Both hURI-tetOFFHep mice and their littermates lacking hURI expression were used in the study, here referred to as hURI(KI/KI)Hep and hURI(+/+)Hep mutants and controls, respectively. Mice were born according to the Mendelian ratio.
Mouse conditions and housing
Both females and males were used in the study, and littermates were randomly assigned as experimental or control groups. All mice have been backcrossed to C57BL/6 for at least seven generations and housed in pathogen-free conditions at the animal facility of the Spanish National Research Centre (CNIO). Animals were maintained within 12 h light/dark cycle between 8:00 a.m. and 8:00 p.m. in a temperature-controlled room in individually ventilated units (22 ± 1°C). Access to water and food was provided ad libidum. Mice were kept in close observation and sacrificed upon signs of sickness, pain or distress following the guidelines for human endpoints for laboratory animals used in biomedical research. All animal procedures have been approved by the CNIO-ISCIII (Instituto de Salud Carlos III) Ethics Committee and Community of Madrid (CAM) and performed in accordance with the guidelines for ethical conduct in experimental animal care and use of the European Union.
Mouse diet and treatment
Mice were fed with a standard chow diet (Harlan Laboratories and Research Diets Inc, #2018S) composed of 18% fat, 58% carbohydrates, and 24% proteins. Food and water were provided ad libidum.
Mouse genotyping
For genotyping, DNA was extracted by incubating mouse fingers with 500 µL of cell lysis buffer containing 1% SDS, 100 mM NaCl, 100 mM EDTA, 50 mM Tris pH 8, and 400 µg/mL of proteinase K overnight. DNA obtained after saturated salt precipitation was further precipitated using 0.7 volumes of ice-cold isopropanol. DNA pellet was washed with 70% ethanol. Purified DNA was dried and resuspended in 500 µL of distilled water. Universal PCR program was used: initial 5 min denaturation step at 94°C, then 35 cycles of 94°C for 30 s, annealing 60°C for 45 s and extension at 72°C for 90 s followed by a last extension step for additional 5 min program. 1 µL of DNA was used for genotyping with primers listed in Extended Table S1.
Temperature exposure for mice
After weaning, hURIHep mice were housed either at room temperature (RT) (23±1°C) (control group) or transferred to the Memmert HPP-life climate chamber (Memmert, #HPP750) for chronic-cold exposure treatments (5±1°C) (treated group). Unless otherwise stated, the treated group was exposed to an acclimatization period (which allowed them to stabilize in their new environment). For this, mice were housed at 18°C for one week, and then after, mice were exposed to cold for 18 weeks and sacrificed at 21 weeks of age for histological analysis. Mice were monitored every day. Notably, independently of the ambient temperature, all mice were housed in 12 hours light-dark cycles, with regular 75% humidity.
Body temperature and weight measurements
Between 3 to 5 pm, mouse rectal temperature was measured using a rectal probe, and mice body weight was recorded using an analytical balance. Mice temperature and weight were monitored weekly, since weaning, and until the experimental endpoint.
Tissue preparation
Mice were sacrificed using CO2 chambers. Full body necropsies were performed, and tissues were removed and weighed using an analytical balance. Macroscopical tumors were counted from freshly harvested livers as described 5, 6, 12. Tissues samples were obtained, washed with PBS, and either kept at -80°C for protein extraction or were fixed in 10% formalin, incubated overnight at RT, and processed for paraffin embedding.
Hematology analysis
After sacrifice, blood was collected from the heart, transferred to EDTA-containing tubes, and kept at 4°C in the dark for direct analysis. To measure glucose, 100 µl of blood were applied in a VetScan mammalian liver profile reagent rotor system (Abaxis, #500-1040) and read in the VetScan Chemistry Analyzer (Abaxis, #D4174-34). The remaining blood was used for plasma collection by centrifugation (4000 rcf, for 20 minutes at 4°C) and kept at -80°C.
Tumor quantification and histopathological scoring
Liver macroscopical tumors were manually counted, and tumor size was measured with image analysis tools available in Fiji software (Image J 2.1.0/1.53c). In addition, histopathological characterization was performed based on hematoxylin and eosin (H&E) staining of liver paraffin-embedded sections with the expertise of the pathologists and co-authors Eduardo J. Caleiras and Cristian Perna.
NAD+ measurement
The total NAD+ levels in mouse tissue were determined using the commercial Kit EnzychromTM NAD+/NADH assay following the manufacturer’s instructions (BioAssay Systems, #E2ND-100) as previously reported 5. Briefly, the kit is based on the lactate dehydrogenase cycling reaction in which the formed NADH reduces a formazan (MTT) reagent. The intensity of the reduced product color (measured at 565 nm) is proportional to the concentration of NAD+/NADH.
Cell culture and treatments
U2OS are human osteosarcoma cells derived from a 15-year-old female. Huh-7 and HepG2 are hepatocellular carcinoma derived cell lines. Cells were cultured in DMEM (4.5 g/L glucose) (Sigma-Aldrich, #D5796) supplemented with 10% Fetal Bovine Serum (FBS) (BioWest, #S1819-500), 100 units/mL penicillin and 0.1 mg/mL streptomycin (Biowest, #L0018-100), 1% glutamine (Gibco, #25030081) and 1% sodium pyruvate (Lonza, #13115E). Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 unless stated otherwise.
Temperature exposure for cells
Before any in vitro procedure, cells were exposed to an acclimatization period where cells were plated and allowed to attach overnight at 37°C. The following morning, cells were washed with Phosphate Buffered Saline (PBS) (Solmeglas, #SBC0300) and fresh pre-warmed medium was added. The temperature exposure in vitro consisted in exposing cells to different temperatures by placing them into three different incubators, maintained either at 27°C or 37°C, importantly a humidified atmosphere of 5% CO2 was always maintained. After temperature exposure, cells were harvested, counted, washed and cell pellets were collected to be processed for experiments as further described, fixed or frozen at -80°C.
Cell number
To determine the effect of temperature on cell number, cells were exposed to different temperatures (27°C or 37°C), as previously described, and harvested at different time points from 2 to 48 hours. After each time point, cells were washed with PBS, trypsinized with 1% trypsin-EDTA (Gibco, #10779413) and manually counted using a Neubauer chamber and Trypan blue (1:10) (Gibco, #15250-061).
Flow cytometry
In order to study the cell cycle, Propidium Iodine (PI) staining was performed. After 24 hours of temperature exposure, cells were harvested. Notably, the medium was not discarded to avoid losing dead cells. Cells were centrifuged and washed three times with PBS. The remaining cell pellet was fixed by adding drop-to-drop ice-cold 70% Ethanol while vortexing. Afterward, cells were incubated for 30 minutes on ice. Thereafter, cells were washed two times with PBS and centrifuge at 2000 rpm for 5 minutes. Cells were then treated with 50 µL of 100 µg/mL ribonuclease (RNAse solution, Sigma, #R6148), and 200-300 µL of 50 µg/mL PI was added. Using the BD LRS Fortessa Flow Cytometer (BD Biosciences) and the BD FACSDiva v9.0 Software, forward scatter (FS) and side scatter (SS) were measured to identify single cells. PI was measured with a maximum emission at 620 nm, using the blue (610/20) and yellow (610/20) lasers to detect DNA. Flow cytometry data were analyzed using the FlowJo v10.5.3 software.
Protein extraction
Cell pellets or mouse tissue was resuspended into 40 to 50 µL of NP-40 lysis buffer (50 mM Tris-HCI (pH 7.4) solution, 150 mM NaCI and NP-40 0.5%) supplemented with protease and phosphatase inhibitors (Complete Mini, Roche #11697498001; Phosphatase Inhibitor Cocktail 2 and 3, Sigma, #P5726, #P0044), and incubated for 30 minutes. Thereafter, lysates were homogenized using the Precellys 24 Bead Mill homogenizer (Bertin Technologies) (15 x 2s, 5500 w) and centrifuge for 20 minutes at 4°C at 16100 rcf as previously reported 35, 36.
Lysate preparation
Lysates were prepared once the same amount of tissue/cell protein extract (25-30 µg) was estimated. To achieve a 25-30 µg concentration, the appropriate amount of protein lysate and the estimated 4X loading sample buffer (NuPAGE, Invitrogen, #NP0007) and 20X reducing agent (Bio-Rad) was added. Next, samples were boiled for 5 minutes at 95°C, spin, and put on ice. Samples were used immediately or stored at -20°C.
Protein quantification
The Bradford method was used to quantify protein concentration. Bio-Rad Bradford reagent (Bio-Rad, #500-0205) and Bovine Serum Albumin (BSA) (Merck, #A7906) was used at different concentrations to perform the standard calibration curve.
Polyacrylamide gel preparation
25-30 µg of protein lysate was added to each SDS-PAGE gels sunk in 1X Running buffer and ran first at 50 V for 20 minutes, and then at 150 V until ready. Transfer buffer was prepared by adding 720 mL of water, 80 mL of 10X Transfer buffer and 200 mL of Methanol. Nitrocellulose transfer membrane (Thermo Fisher, #88018) and Whatman 3 MM paper (Sigma-Aldrich, WAA30309117) were pre-wet in Transfer buffer. Subsequently, the transfer “sandwich” was assembled in a wet blotter in the following order: anode- sponge- 2 Whatman papers- Gel- Nitrocellulose membrane- 2 Whatman papers – sponge- cathode. Bubbles were removed, and the gel was transferred for 2 hours at 100 V. When finished, membranes were washed with water, and the protein transferred was checked by staining with Red Ponceau solution (Sigma-Aldrich, #6227-79-5).
Western blot analysis
Membranes were washed and blocked with 5% blotting grade blocker dry milk (Bio-Rad, #1706404) dissolved in Tris-buffered saline containing 1% of Tween-20 (PanReac, #A4974) (TBS-T) for 1 hour at RT. Subsequently, membranes were incubated with the corresponding primary antibody diluted either in blocking solution or TBS-T-BSA (5% BSA in 1X TBS-T). Membranes were incubated overnight at 4°C in agitation. After incubation, membranes were washed three times with 1X TBS-T for 10 minutes and incubated with the corresponding secondary antibody directed against mouse or rabbit IgGs (Dako, goat anti-rabbit #P0448, goat anti-mouse #P0447) for 1 hour at RT in agitation. Finally, the membrane was washed twice with 1X TBS-T for 10 minutes and immunoblots were incubated in 1:1 homemade ECL composed of Solution A (1.25 mM luminol, 0.2 mM p-coumaric acid in Tris-HCL 0,1 M (pH 8.5) and Solution B (0.03% H2O2 in Tris-HCL 0.1 M (pH 8.5) for 1 to 5 minutes. Lastly, immunoblots were developed in X-ray films (Fujifilm, #47410) for protein visualization.
Hematoxylin and Eosin
For histological studies, hematoxylin and eosin (H&E) staining were performed on paraffin-embedded sections. Tissue sections were deparaffinized in xylol, rehydrated through graded washes of Ethanol and washed with distilled H2O. Slides were stained with hematoxylin for 10 minutes and rinsed with distilled H2O. Subsequently, slides were counterstain with eosin for 2 minutes and washed. Slides were then dehydrated successively with 96% and 100% Ethanol, washed, and mounted with xylol.
Immunohistochemistry
Immunohistochemistry (IHC) was performed in 3 µm paraffin-embedded sections as previously described 37, 38, 39. Tissue sections were de-waxed, rehydrated and subjected to heat-induced antigen retrieval in 1 M sodium citrate buffer (pH 6). For blocking endogenous peroxidases, samples were incubated for 10 minutes with 3% H2O2. Subsequently, samples were permeabilized in 0.2% Triton X-10 in PBS for 20 minutes, and sections were then blocked for 1 hour at RT with 5%(rabbit/horse/goat/rat) serum in 0.2% Triton X-100 in PBS. Sections were then incubated overnight at 4°C with their corresponding primary antibodies. Sections were washed with PBS three times, and the Vectastain ABC Kit (Vector Laboratories, anti-rabbit #PK-4001, anti-mouse #PK-4002, anti-goat #PK-4005, anti-rat #PK-6104) was used following the manufactures instructions. After that, sections were developed using 33-diaminobenzinetetracloride chromogen (DAB) (Dako, #K3468) and counterstained with hematoxylin. Lastly, sections were mounted with xylol.
Immunofluorescence
Immunofluorescence (IF) in cells was performed using a 384-well Cell Carrier black plate with an optically clear bottom (PerkinElmer, #6007550) or in glass coverslips placed on 12-well cell culture plates as previously reported 38, 40. Cells were exposed to temperature exposure treatments as previously described. After that, cells were washed with PBS and fixed in 4% paraformaldehyde (PFA) (ProSciTech, #C004) for 10 minutes on ice. Then, cells were washed with PBS and permeabilized with freshly prepared Triton 0.5% in PBS for 20 minutes. Cells were then blocked for 30 minutes in a blocking solution consisting of 5% BSA and 0.5% Triton in PBS. Subsequently, cells were incubated for 2 hours with its corresponding primary antibody diluted in blocking solution. After washing, secondary antibody incubation with anti-rabbit, anti-mouse IgG antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 555 (Life Technologies, goat anti-mouse Alexa Fluor-488 #A11001, goat anti-mouse Alexa Fluor-555 #A21422, goat anti-rabbit Alexa Fluor-555 #A21429) and DAPI (1 µg/ml) (Merck, #D9542) was performed in 1% BSA PBS-T for 1 hour, at RT. When using 384-well plates, plates were washed for 5 minutes, three times and 100 µL of PBS was added to each well. Images were automatically acquired from each 384-well using the Opera High Content Screening System (Perkin Elmer). When using glass coverslips, slides were mounted using Prolong Gold Antifade Reagent (Thermo Fisher, #P36934) allowed to dry overnight, in the dark at 4°C. Images were acquired using a Confocal Ultra-Espectral Microscope SP4 (MP) (Leica Microsystems). Three replicates of each condition were used in every independent experiment, and every condition was analyzed in at least three independent experiments.
Image analysis
For image analysis and quantitative measurements, a minimum of 10 images per slide/well were acquired at a 10X-20X magnification. IHC quantification was performed by manually counting the number of positive cells per area in a minimum of 10 microscopic field images using the Cell Counter plug-in in Image J Fiji software (Image J 2.1.0/1.53c). For IF, images were manually quantified or analyzed with the help of the CNIO Confocal Microscopy Unit using Definiens Developer v2.5 or the Opera Screening System.
Statistical analysis
Statistical analyses were performed using GraphPad Prism v6-9 software. Statistical significance (p) (p 0.05 = *, p 0.01 = ** p 0.001 = *** and p 0.001 = ****) between the means of a minimum of three groups was determined using unpaired two-tailed Student’s t-test or linear regression analysis. Unpaired two-tailed Student’s t-test was employed for comparisons under normal distribution, otherwise, its non-parametric alternative, the Wilcoxon-Mann-Whitney test was applied. Shapiro-Wilks normality test was performed to assess normality. ANOVA allowed comparing more than two groups when normality assumptions were met, otherwise, the non-parametric Kruskal-Walli’s test was employed. Survival was analyzed using Kaplan-Meier estimators. Results are expressed as the mean value ± Standard Error of the Mean (SEM). ‘‘n’’ represents the number of mice or experiments used in each experiment, as indicated in Figure Legends. All results are representative of at least three independent experiments.
Data and materials availability
All data are available in the main text or Methods. Materials are available upon request to N.D. and the sharing of materials described in this work will be subject to standard material transfer agreements.
REFERENCES
For full bibliography please see the pdf file.
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
AUTHOR CONTRIBUTIONS
We thank all mouse providers as described in Methods. We are also thankful to the CNIO Mouse Genome Editing Core Unit and Animal Facility for the mouse re-derivation and maintenance, respectively. This work was funded by grants to N.D. supported by the State Research Agency (AEI, 10.13039/501100011033) from the Spanish Ministry of Science and Innovation (PID2021-122695OB-I00), also including the iDIFFER network of Excellence (RED2022-134792-T), cofunded by European Regional Development Fund (ERDF) and by AECC (PRYGN211184DJOU). This work was developed at the CNIO funded by the Health Institute Carlos III (ISCIII) and the Spanish Ministry of Science and Innovation.
ACKNOWLEDGMENTS
We thank all mouse providers as described in Methods. We are also thankful to the CNIO Mouse Genome Editing Core Unit and Animal Facility for the mouse re-derivation and maintenance, respectively. This work was funded by grants to N.D. supported by the State Research Agency (AEI, 10.13039/501100011033) from the Spanish Ministry of Science and Innovation (PID2021-122695OB-I00), also including the iDIFFER network of Excellence (RED2022-134792-T), cofunded by European Regional Development Fund (ERDF) and by AECC (PRYGN211184DJOU). This work was developed at the CNIO funded by the Health Institute Carlos III (ISCIII) and the Spanish Ministry of Science and Innovation.
COPYRIGHT
© 2024

Cold exposure reinstates NAD+ levels and attenuates hepatocellular carcinoma by Grazioso et al. is licensed under a Creative Commons Attribution 4.0 International License.


